
��ѯ���ߣ�
17715390137
18101240246
18914047343
�ʼ���mxenes@163.com
ɨ���ע�����������ںţ�
������Fronrier
��ע�������½���ϵ���ǣ�
������ҵ�š�
רҵ��������


DOI: 10.15541/jim20230123
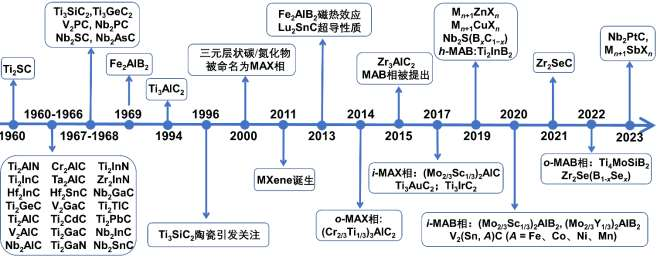 ͼ 1 ��Ԫ��״���ϵķ���ʱ���� (����ȫͳ��)
ͼ 1 ��Ԫ��״���ϵķ���ʱ���� (����ȫͳ��)
Part1
�� ������������з�ƽ̨�������ھ� ��
�ڽṹ����MAX ��ȫ������������ϵ���� MAB �ಿ������������ϵ����������������ϵ�����������MAB ���� B ԭ���� M ԭ�ӵı���Ҫ���� MAX ���� C/Nԭ���� M ԭ�ӵı�������ˣ�MAB ��û������ MAX ������ƣ�Ӧ��ע�����ߵ����֡�MAB �����˽������մɵ����ԣ������������ѧ���ܣ�Ϊ��þ��ж������ܵĸ�ǿ�����ṩ�˹���ǰ����ͨ����һ��ԭ�����㣬MAB �����ѧ���ʣ������ģ����B��������ģ����G��������ģ����E���Ͳ��ɱȣ��ͣ��ȵõ��㷺�о������3 ��ʾ[160, 164-168]�����У�TcAlB��Nb2AlB2��W2AlB2��Tc2AlB2 �� Co2AlB2 �� Ni2AlB2 Ϊ���Բ��ϣ���Cr2AlB2��Mo2AlB2 �� W2AlB2 ���и�ģ������Ӳ�Ⱥ͵͵��Ը������ԣ�������ʵ�ʹ���Ӧ�á�
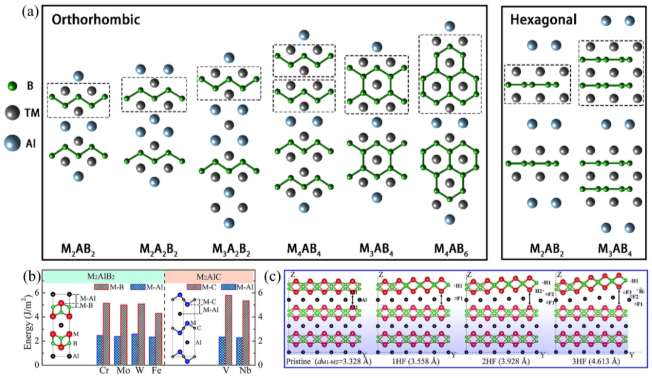
ͼ 2 MAB ������ۼ���





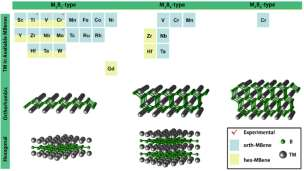
ͼ 3 MBene �����༰�ṹʾ��ͼ
Part2
�� ���ϻ������ͨ������ɸѡ ��
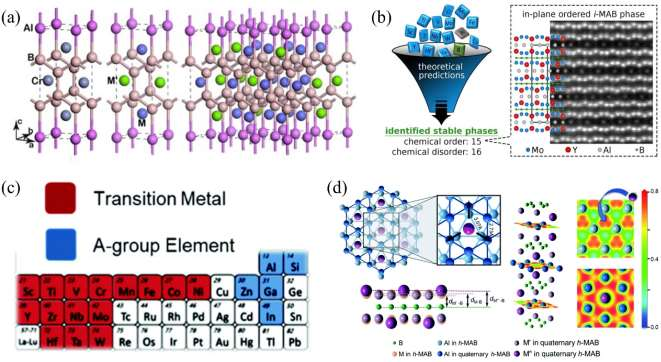
������֪��MAX����һ���������ɽ�����״̼/�����������Ԫ�ض�������̼���ϣ���δ���̼��Ԫ�ص����ƣ��� MAX ���Լ� MXenes ������Ԫ����չ�����������о��ߵ�һ�����⡣2019 �꣬�����ܵ�[52]ͨ�����ϻ��̷������ɹ�ͻ������һ���⣬�״�����Ԥ�Ⲣʵ��ϳ��˵�һ������������ϵ����Ԫ���ɽ������Ti2InB2��ͼ 14(a)�����������Ȳ��ö�Ԫ��ɷַ�ȷ���� TixBz���ȶ��Ժͽṹ��Ȼ���� TixBz�� AΪ��ʼ��Ԫ������α��Ԫ�ṹ�����������õ�����ѧ�ȶ��� Ti2InB2�ṹ����� Ti2InB2չ����Ԫ��ɷ���������ȷ������Ԫ�����ȫ���ȶ��ԡ�����һϵ�еļ�����ɸѡ������Ti2InB2 �ڳ�ѹ�¾���ȫ���ȶ��ԡ���һ���أ������ܵ�ͨ�����෨�ɹ��ϳ��˲�״���� Ti2InB2���������������ɽ�����״����ֻ������̼������һ���ޡ����������[162]����֪���͵�MAX ���Լ� Ti2InB2 Ϊ���ӣ�ͨ����ͨ�������������� Zr-AB �� Hf-A-B ��ϵ�з�����һϵ�о�������ѧ�Ͷ���ѧ�ȶ��Ե���Ԫ��״���ɽ���������������ġ�212�������⣬�����־��������¹��͡�211���͡�314������Ԫ����Ҳ�����ȶ����ڡ��ù�����һ�������������Ԫ��״���ɽ���������ȶ����ƣ��� B�C B �������ʯīϩ״���ڡ�212���͡�314�����������ȶ����ã�����211�������� MAX ��Ľṹ���ƣ��� MB6 �������ǽṹ���ȶ���Ԫ����ͼ 14(b)�����⣬��ά����[183]���ܶȷ�������������㷨���ϣ��� P��S��ϵ��Ԥ�����߶���ѧ������ѧ�ȶ��Ե�Nb2PB2��Nb2PB �� Nb2SB��ͼ 14(c)������Щ��Ԫ��״�մɱ��ֳ�������ȵ����ܣ����� Nb2PB2���ȵ��ʸߴ�-65 W��m-1��K-1���ܵ���˵����Ԫ�ػ�ѧ��������������ˡ�211�����ͷ��ϴ�ͳ�� Mn+1AXn����ʽ����212���͡�314�������������� MAB �ࣨorthorhombic MAB phase�� ��� ort-MAB����Ԫ�ر�����Ϊ�˸��õ����������������ṹ�� M-A-B Ԫ����ɵ���Ԫ�����������Ϊ���� MAB �ࣨhexagonal MAB phase�����Ϊ h-MAB �ࣩ��
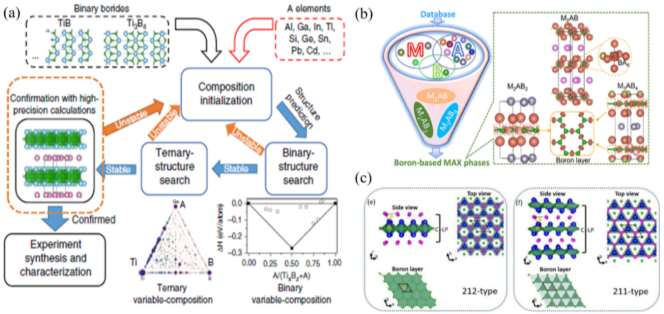
ͼ 4 ��Ԫ MAX ��ķ���
ͼ 5 ��Ԫ MAB �������Ԥ��
Part3
�� �ṹӳ��������ȶ����о� ��
������Ͻṹ��Ҫͨ��������Խ���ȷ����1��ʵ��������÷���ʼ�ڷ롤�Ͷ�[190]��������ȵ����ڹ�����������Ϊȷ�����Ͻṹ�ĺ��IJ��ԣ�Ȼ�������������˸�ͨ�����Զ������ֶΣ��÷�����Ȼ���к�ʱ�������Ȳ��㡣2�����㽨ģ�����Ÿ��ټ�����ķ�չ���Լ�����������ý��мĵ������Ƽ��㷽���ķ�����ʹͨ��������ѧԭ���������Ԥ����Ͻṹ��Ϊ���ܡ�Ȼ�����ò���Ŀǰ��Ȼ������һЩ���ѣ����������i������Chelikowsky���ᵽ����Ȼ���ǶԻ������д��ڵ�ԭ�Ӽ�������Ѿ����˳����ʶ�����ǶԹ������������ļ�����Ȼ�dz����ѡ�������Ϊ��ԭ�ӵ��ܼ����� 10 6 eV ���𣬶�����ܽ�����1~10eV/atom �ķ�Χ�ڣ��ɴ����DZ������ 0.000001 ����ߵļ��㾫�Ȳ��ܶԴ�������ȷ�Ĺ��㡣ii��������ԭ�ӡ����Ӻ͵��ӵ���Ŀ���Ӿ��˴��������Ѷȣ��Ӷ�ʹ�÷�����Ԥ�⾫���ϵ���ʵ��۲⡣3���ṹӳ�����ò��Ի������еIJ��Ͻṹ���ݣ���ϻ�ѧ��Ԫ����ز�����������ֵĿ��ܽṹ���й�����Ԥ�⣬���ڴ˻������ܽ���γ��ض��ṹ��һЩ���ɺ�ģʽ���÷��������������궨�����������Խṹ���ݽ��з��࣬�����ݿ��еĴ����ṹ����ӳ��ͼ�еIJ�ͬ������л��֣����Դ�Ԥ������ֵĿ��ܽṹ���ͣ��ṩѰ��������Ͻṹ�ij�ʼ���Ӳ�����Ϣѧ�Ĺ۵��������ṹӳ������һ�ֶԾ���ṹ���о�������ķ������ṹӳ������������Mooser-Pearson ӳ�䡢Zunger ӳ�䡢Villars ӳ���Լ���Ϊ������Pettifor ӳ�䡣
ͼ 6 ѡȡ������Ϊ����Ũ�Ⱥͳߴ����������ƵĽṹӳ��ͼ
 |
 |
 |
 |
| ��ά����Frontier | �������ײ���ǰ�� | MXenes Frontier | ����ҽѧFrontier |

| ��ܰ��ʾ�����������²ĿƼ�����Ӧ��Ʒ�����ڿ��У������������塣������վʾ��ͼԴ�Ի�������ͼƬ�����ο�������ʵ�ʲ��Խ��Ϊ��������Ȩ����ϵ��������ɾ������Ʒ���������ο�������ʵ��ֵΪ�� |
|
��Ȩ���� © 2019 ���������²ĿƼ�����˾
All rights reserved. ��ICP��16054715��-2 |
ɨһɨ