
��ѯ���ߣ�
17715390137
18101240246
18914047343
�ʼ���mxenes@163.com
ɨ���ע�����������ںţ�
������Fronrier
��ע�������½���ϵ���ǣ�
������ҵ�š�
רҵ��������

���������
��������ӵ�ر��㷺Ӧ���ڱ�Яʽ�����豸�͵綯�����У�Ŀǰ��Լ��������ӵ��Ӧ�õ�ƿ��������DZ�ڵİ�ȫ�������봫ͳ�����ʹ�õ���ȼ���ױ���Һ̬�л������ϢϢ�йء��з��������̬�����(SSE)Ϊ������ȫ��̬����ǽ��DZ�ڵ�ذ�ȫ���������Ч;����Ŀǰ��̬�������ϵ���·�Ϊ�����ࣺ���������ʡ��������ʡ����������ʾ��нϺõ��ȶ��ԡ��Կ�����ˮ�ֲ����С�ͬʱ���нϿ��ĵ绯ѧ���ڵ����ơ���Ҳ�������ӵ絼�ʽϵ͡�����/��������ϡ��״�ϡ��Ʊ��ս��¶Ƚϸߵ����⡣�������ʾ߱����ߵ����ӵ絼�ʡ��õ���չ�ԡ�С�ľ���/�������衢�ϵ͵��Ʊ��ս��¶ȵ�����������̰�ͬ�����ԣ����磺��ѹ����խ�����ѹ�������ϼ����Բ�����Li�����ȶ�����⡢�Կ�����ʪ�����еȡ�̽Ѱ�ۺ����ܸ�����������Ϳ����ӵ����Ϊؽ�����Ĺؼ�����֮һ�����⣬ȫ��̬����еĹ�-�̽�����ȶ��ԡ������ԡ�ƥ��ȵ����������Ȼ������Լ��ȫ��̬��صĵ绯ѧ���ܡ�
���Ӵ�����ݿ��У���������Ļ�ѧ�ɷ���ϣ�Ѱ������̬����ʵ���ϵ������ͨ��ʵ���Դ����ѿ��پ���Ѱ�ҵ���������ĵ�����䷽������һ���棬���ȫ��̬����й�-�̽������⣬��Ҫ�����ԭλ���Բ��ܱ����������ͨ�������ܶϺ��Ѱ��չؼ��Կ�ѧ���⡣��ˣ�ؽ��һ�۵��о��������������͵���ʵ�̽Ѱ����������Ϊ����һ�廯ȫ��̬����ṩ������������֣�ݴ�ѧ�۹�ʤ���ڿ����飬ͨ�����ϲ��ϻ����鷽������Թ�̬����еĹؼ����⣬�γ���һ����DFT�㷨�����и�ͨ�����㣬̽Ѱ����̬������Լ��ṩ���ȫ��̬��صĹ̹̽�������Եĺ����Է�����Ϊ����ʵ���з��ṩ������������֧�š�
���ɹ���顿
֣�ݴ�ѧ�۹�ʤ�����о��Ŷ���Energy Environ. Mater�Ϸ�����Ϊ��First Principle Material Genome Approach for All Solid�\State Batteries�������¡�
��ͼ�ĵ�����

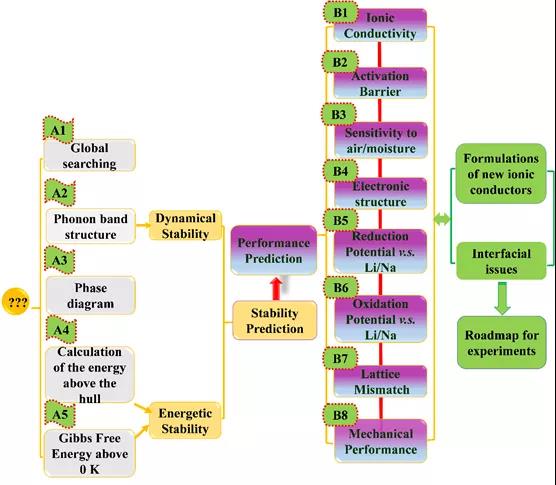
ͼ 1. ���ϻ����鷽�����̡�
��ͨ�����ϻ����鷽���������̣���ͼ1��ʾ����������A������ѧ�Ͷ���ѧ�ȶ���Ԥ�⣬��B����̬����ʹؼ�����Ԥ�⣬���֡�(A)���ֺ��ǵIJ��ϻ����鷽��������USPEX�Ŵ��㷨�����Ӵ����㣬��ͼ������0 K�γ��ܼ��㣬���¼���˹�����ܼ��㡣������Ʋ��ϵĽṹ�ȶ��Ժ�����ѧ�ȶ��Գ��Ԥ������������Ʋ����ں��������еĿ�ʵʩ�ԡ�(B)���֣��������ӵ絼�ʡ���ɢ�����ܡ����ӽṹ��������ԭ��λ������ʧ��ȡ���ѧ���ܵȹؼ����ܽ��п�Ԥ�⣬��ͨ��̽Ѱ����̬����ʡ�����Թ�̬����еĹ�-�̽������⣬��ߺ���������������֧�š�
���������Li6PA5X����ʵ�������ƺ�ʵ���Ʊ���
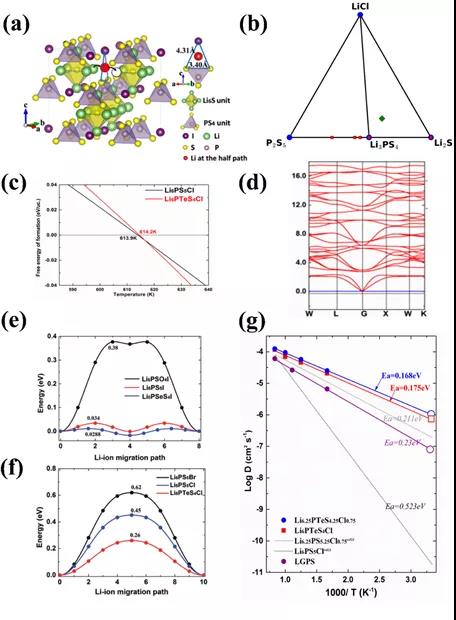
ͼ2. (a)Li6PS5X�е�Li+������ɢͨ��������ƿ����(b) Li6PS5Cl��������ͼ��(c)ͨ������˹�����ܼ���ó���ת���¶ȡ���d��ͨ�����Ӵ�����Ԥ��Li6PTeS4Cl�Ľṹ�ȶ���(e)����f������CI-NEB���㷽������ȷ���Է������������ɢ�Ķ�Ӧ��ϵ����g��ͨ��AIMD����Ԥ�����������������
���������Li6PS5X����ʹ����У�����S-X-S�����ε�ƿ��������ͼ2a��ʾ�������������������ӵĿ������ˡ��ʣ�ؽ��ͨ�����ԣ���ͨ��������ƿ������ʵ��������ӵĿ������������Ĺؼ��������ֹ��������������͵���ʣ��ṩ�������������������ȶ��Ժ��������������������Ԥ�����������£�ͼ2��b������ͼ����������Li3PS4��LiCl, Li6PS5ClΪ�����ࣻͨ������˹�����ܼ��㣨ͼ2��c���������ֵ��¶ȸ���614Kʱ��Li6PS5Cl��ת��������࣬Ϊ����ʵ�鹤���ṩ�ȴ���������ͨ�����Ӵ����㣨ͼ2��d����,����Ƶ˵����ṹ�ȶ��������ȶ���˵������ƽṹ���ȶ����ᷢ����ת�䡣��Ե���ʵĹؼ������������ܣ���ͼ2��e��-(g)��,CINEB��AIMD�������ϣ���������ѡ��Ѹ��Է�����ͨ���ȶ����ӵ絼�ʺ���ɢ�����ܣ���ѡ����������䷽Li6.25PTeS4.25Cl0.75��Te����ȡ��Sλ��������������ӵ���ɢƿ���������������������͵���ʵ����������������
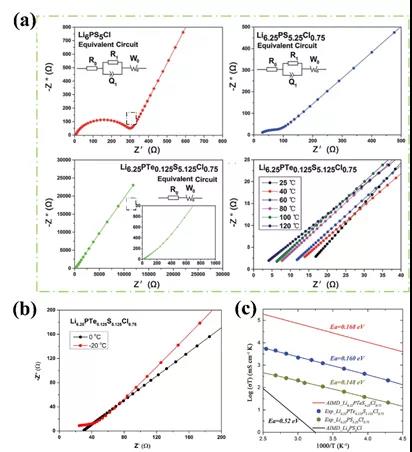
ͼ3.��a��Li6PS5Cl,Li6.25PS5.25Cl0.75, Li6.25PTe0.125S5.125Cl0.75�����µ绯ѧ�迹ͼ�ס��Լ����µ绯ѧ�迹ͼ�ס�(b) Li6.25PTe0.125S5.125Cl0.75���ڵ��»����µ绯ѧ�迹ͼ�ס�(c) Arrhenius ��ϵlog(��T) vs. 1000/T��ʵ���������Ƚϡ�
��ǰ��������Ƶ�ָ���£����ǿ�����ɹ��Ʊ�����Li6.25PTeS4.25Cl0.75����������ʡ���Te����ȡ��S����ͼ3��a����ʾ�����ӵ絼�ʵ�����������ߡ�Li6.25PTeS4.25Cl0.75���������ӵ絼�ʸߴ�4.5 mS cm-1������ɢ����С����Ϊ0.160 eV�������ڳ����»����£�-20�棩������ӵ絼����Ϊ1.612 mS cm-1����ͼ3��b����ʾ��AIMDԤ����������ɢ������Ϊ0.168 eV��ʵ������0.160 eV��ȫ��Ǣ���磬ͼ3��c����ʾ��
˫�����ѿ�������ϵ��������ƺ�ʵ���Ʊ���
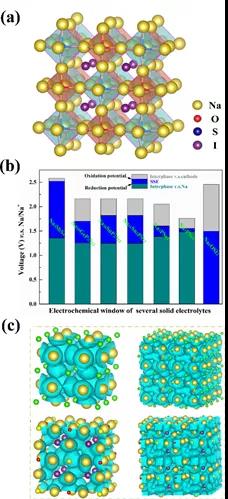
�������е�����������Խ���Li��Na������������صĸ���Ӧ���ᱻ�ֽ��Li3P, Li2S, Na3P, Na2S���������ӵ��塣ؽ��һ�������Li��Na�����ļ����������ĵ������ϵ����֪�ĵ��ͷ����ѿ�����Li3OCl, Li3OBr,Na3OBr, Na4OI2���нϺõ������Li��Na�����ļ����ԣ����������ӵ絼�ʽϲ��˻��ڲ��ϻ����鷽�����ڵ������ѿ�ṹ�Ļ����ϣ�ͨ���ṹ���ԣ���Ʋ��Ʊ�����˫�����ѿ����ʡ�
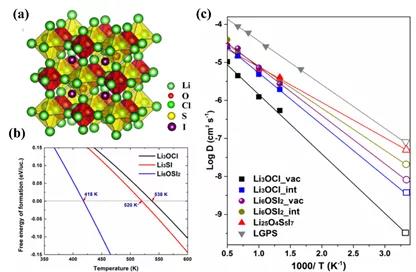
ͼ4. (a) Li3O0.5S0.5I�ľ���ṹͼ��(b) Li3O0.5S0.5I�ļ���˹�����ܡ�(c) ͨ��AIMDģ���������ɷֵ�﮵�������������ɢ�����ܡ�
˫�ͷ����ѿ�Li6OSI2�ľ���ṹ����ͼ4(a)��ʾ������ռ��λ���γ�˫�ͷ����ѿ�Li6OSI2�ṹ��ͼ4 (b)ͨ������˹�����ܼ���Li6OSI2ת��Ϊ�ȶ�����¶�ת���Ϊ418K��ͼ4 (c)ͨ��AIMD����ı�����Ӧ���������ӵ���������,��ȷ�����߱����ŵ���������������
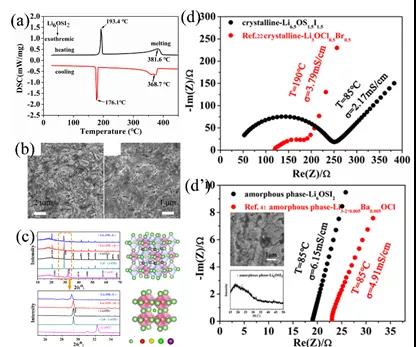
ͼ5. (a) ˫�ͷ����ѿ�Li6OSI2��DSC���������ߡ�(b)�Ʊ��մ������ͺ�����ɨ��ͼƬ��(c)ʵ��XRDͼ�������XRDͼ�ס�(d)����Li6.5OS1.5I1.5���迹ͼ�ס�(d��)ͨ���Ǿ������������������迹ͼ��
����DSC������ͼ5 (a)��Li6OSI2ת��Ϊ�ȶ�����¶���176.1�棬������Ԥ��418 K ��145 �棩����һ�¡�ͨ���ս��ȴ������õ�Li6OSI2����, ͼ5 (c)��ͨ���Ǿ�����ͼ5 (d��),���Լ�������/�����迹��Ӱ�죬�������������������ڵ��ͷ���̬�����ʡ�
ͼ6. (a)Na3O0.5S0.5I˫�����ѿ�ṹ(b) Na3O0.5S0.5I���ȶ���ѹ����(c)���Ӷ���ѧNa+�������˹켣��
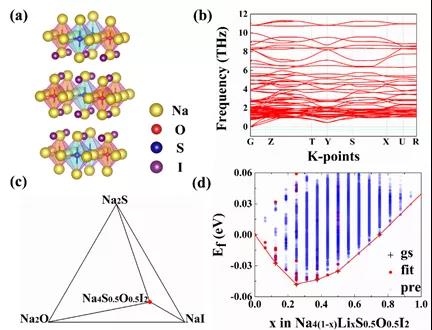
ͼ7.��a��Na4S0.5O0.5I2��״�����ѿ�ṹ����b��Na4S0.5O0.5I2������������c��Na4S0.5O0.5I2��ƽ����ͼ����d��ͨ��ATAT�Ŵ�չ��������Na4(1-x)LixS0.5O0.5I2�ɷ���ͼ��
���ڲ��ϻ������ͨ���㷨��������Ƴ�Na3O0.5S0.5I˫�ͷ����ѿ�(ͼ6)��Na4S0.5O0.5I2��״�����ѿ�(ͼ7)���������ӵ������ϵ��������ɷ־���ͨ�������ȶ��ԺͶ���ѧ�ȶ��Եı��������ܷ��棬���ƻ�ѧ�����ȶ����߱�������������������������ͼ8��ʾ���Բ�״�����ѿ�Na4S0.5O0.5I2����һ�廯ȫ��̬�����ӵ�ز��ԣ��ȿ��Գ�Ϊһ�����Ҽ��ݵIJ���ϵͳ���ֿ���Ϊ���õĵ���ʺ��������������ܶȳ���320 wkg-1�����ʵ�����ڿ�չ�С�
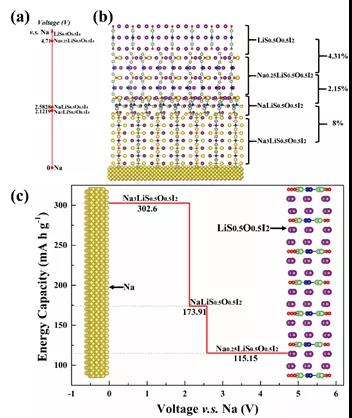
ͼ8. ��Na4S0.5O0.5I2Ϊ����������һ�廯������ϵͳ��
�������������Li6PA5X����ʵ�ȫ��̬���һ�廯���ԣ�
�������缫�ļ����ԣ�
����������ϵ�������������ӵ絼�ʺ���չ��ʹ���ڹ������о��й�����Ӧ��ǰ�����������������̬����ʵĵ绯ѧ����խ����ߵ�ѹ��������ֱ�ӽӴ�ʱ�������ڽ��洦�γ�����ӵĺľ��㡣���ң�����������Li������������ڽ�Ϊ���صĸ���Ӧ��������谭������������ȫ��̬����е����á�

ͼ9.��a��ͨ��AIMD���㣬����Li6PS5Cl�����Li�����ĸ���Ӧ����b�� ����ƽ����(0K)��DFT���㲻ͬ��������ϵ�ĵ�ѹ���ڡ�(Y. Zhu, X. He, Y. Mo, A. C. S.Appl, Mater. Interfaces 2015, 7, 23685)��c������ATAT�Ŵ�չ������SSE��Ƕ�����﮹��������γɵIJ�ͬ�ṹ��
��ͼ9 (a)��ʾ������ͨ������Li6PS5Cl|Li������ģ�ͣ�����ΪLi6PS5Cl�����Li������ᷢ�����ظ���Ӧ������ʻᱻ�ֽ��Li3P���м���Li6PS5Cl����ʻ�ԭ��λ�ϸߣ�ԼΪ 1.7 V(��ͼ9 (b)��ʾ)������绯ѧ��λ��Li�����ݡ���һ���棬������ʴ��ڸ�ѹ���ʱ��Li6PS5Cl������ʧȥﮣ���������P2S5,LiCl,S���䵼���������������м�����(��ͼ9 (c)��ʾ)��
��ˣ�����������Ե������Li6PO5Cl��Li6PO4SCl���ֹ����Ի������ϣ�(��ͼ10 (a)��ʾ)��ͨ�����㷢�����벿��O��﮵��ȶ��Դ������������ͼ10 (b)��ʾ�����һ���������������ϵ�߱����ƵĻ�ѧ��ɣ������ڱ�������������ʼ䷢�����صĻ�ѧ����Ӧ�����ң��������������ʼ��������ľ���������Li6PO4SCl/Li6PS5Cl/Li6PO5Cl����ʧ��Ƚ�Ϊ1.5%��3.1%�����õľ���ƥ���ϵ���Ͻ��洦��Ӧ��С����������Li0.25MnO2|Li6PO5Cl|Li6PO4SCl|Li����ʽ����һ�廯ȫ��̬��ء�
ͼ10.��a��ͨ��ATAT����O/S�������õ��ĸ��ϳɷ֡���b��) ����ΪLi6PO5Cl|Li��Li6PO4SCl|Li��Li6PO5Cl|xLi6PO4SCl������ģ�͡�����Ϊ����AIMD�ڸ��³�ʱ���ݻ��������ģ�͡���c����Ƹ������ܶȵ�һ�廯ȫ��̬��ء�
�������ܽ
�Բ��ϻ����鷽��Ϊָ�������Dz���DFT��ͨ�����㣬��Բ��������ȶ��ԣ��ṹ�ȶ��������ؼ�Ԥ�У�ָ��ʵ��ϳɡ��������ӵ絼�ʡ���ɢ�����ܡ�������ԭ��λ�ȵ���ʻ����ؼ���������ȫ��Ŀ�ѧԤ����������ʵ�����ϣ�����һ�廯ȫ��̬��ص�ʵ�û��Ų���
�������ӣ�
https://onlinelibrary.wiley.com/doi/full/10.1002/eem2.12053
����չ��
�����ĵ���֣�ݴ�ѧ���Ҽ���Ƹ�����۹�ʤָ������ɣ�ͨѶ���ߣ���֣�ݴ�ѧ����������ʿ�о�������Ϊ��һ���ߣ�֣�ݴ�ѧ������Ȼ��˶ʿ�о�������Ϊ��ͬ��һ���ߣ�֣�ݴ�ѧ�����������ڣ���ͬͨѶ���ߡ��ù�������Ȼ��ѧ����ί��֣�ݴ�ѧ����ѧԺ��֣�������Ͳ��ϻ����鹤���о�Ժ��http://www.zmgi.net/����������ɡ���лEEM������̽�����������ӡ�ͬʱ��Ҫ��л֣�ݡ����������Բ��ϻ���Ժ�Ĵ���֧�֣�ʹ�����ܹ�ͨ��Դͷ���¡���л���۸����ܼ��㣬�Ա������ṩ�ķ��������������֧�֡�
�۹�ʤ�����Ҽ���Ƹ���ڡ�����ְӢ��������ѧ�����о��٣���³�ζ���ѧ����ѧ�����ڣ�Ӣ�������ٴ�ѧ�������ѧ���ڡ�����Դ�о�������������ԺԺ��������������ѧ�����εȣ�Ӣ�����ϻ�ѧίԱ��ίԱ���ɳ�����Դ���Ϲ������Ա��2010����ѡ���ҡ�ǧ�˼ƻ�����������֣�ݴ�ѧ����������Ӣ����ܲ����о����ģ�2012���������϶�Ϊ����ʡ��̼���������Ϲ�������ʵ���ң�2014�� �Ƽ������� ���Ҽ���̼����������������ƹ�������ʵ���ң�2015���Ƽ�������2016���ڡ���ԭ�ǹȡ�����֣�������Ͳ��ϻ����鹤���о�Ժ�������˹����ڿ���Energy & Environmental Materials (EEM)������John Wiley & Sons, Inc���档�о������ڶ�߶Ȳ���ģ�⼰���ܲ�����ơ�������Ĥ�����Ʊ��������Ƚ����ϱ���������Դ�����������ϼ����ȡ�����������Nature�����ڵĹ��������ڿ�����200��ƪ�����벢��ù�����ר�����
��Ϣ��Դ��
 |
 |
 |
 |
| ��ά����Frontier | �������ײ���ǰ�� | MXenes Frontier | ����ҽѧFrontier |

| ��ܰ��ʾ�����������²ĿƼ�����Ӧ��Ʒ�����ڿ��У������������塣������վʾ��ͼԴ�Ի�������ͼƬ�����ο�������ʵ�ʲ��Խ��Ϊ��������Ȩ����ϵ��������ɾ������Ʒ���������ο�������ʵ��ֵΪ�� |
|
��Ȩ���� © 2019 ���������²ĿƼ�����˾
All rights reserved. ��ICP��16054715��-2 |
ɨһɨ