 ��˾���
��˾���
��ز���
QQѧ������Ⱥ��1092348845
��ϸ����
��3����Science��Nature�����IJ�����ع����У��н�70%ʹ�õ��˼���ģ�⡣���ͬʱ���ڸ����������У��������漰����غʹ����������Ĺ���ʱ������ģ���Ѿ���Ϊ��Ҫ�ķ�����֤���ֶΡ�
�Ƚ����ε��ǣ�ʵ�������ĺ蹵��Ȼ�ܴܶ�ʵ����Ա���������ģ���ܽ����Щ���⣬���Լ��о����������Ӧ�ã�
���Ƿ�����5000��ƪ����ģ���ڲ����о��е�Ӧ�����£�ɸѡ�˽�ǧ���ʰ�����������ѡ������121ƪ��ز�����ع������϶��ߡ�
�dz�ֵ��ע����ǣ������������ѡ��121ƪ���㰸���У���Ȼ��72ƪ��ʹ��VASP����������ġ�VASP��Ŀǰ��õIJ��ϼ�����ҵ����֮һ����MedeA��ΪVASP������������һ�����������Ƚ���������ѧVASP�ͷ��Ӷ���ѧLAMMPS�����һ��ƽ̨������������ʵ����Windowsϵͳ�´���ģ�͡����ò��������ӻ�������������ڲ���ʹ�á�
��ز�����ؼ��㹤������Ҫ�漰����10������
1. ��������ģ��������������ض����ϻ���������ܡ�����λ���������ȡ�
2. ��Ӧ����ģ������ͬ�����ܶ��µij�ŵ�����ģ�⡢�缫���ϳ�ŵ��������ת�䡢��Ӧ����̬ģ�⡢������ʧ��������Ӧ��λ�ݱ䡢�����ܡ���ɴ�����Ƶȡ�
3. ������ɢģ����ģ�������ڲ����е���ɢ������絼�ʺ���ɢǨ��·���ȡ�
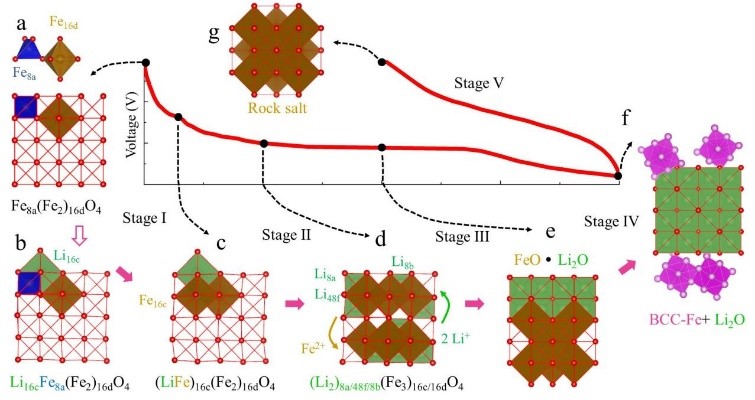
4. ���ϸ���ģ��������/�����Ե��ӽṹ������ṹ����ṹ�����ӵ絼�ʡ����ӵ絼�ʡ������������缫��λ����ѹ���ߡ���·��ѹ��Ӱ��ȡ�
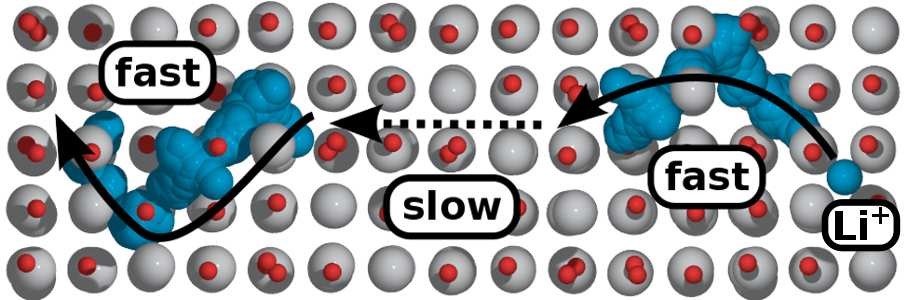
5. ���淴Ӧģ�������Һ/���Ӽ���缫/SEIĤ��������á������֦�������������ƻ������ض�����ķ�Ӧ���Եȡ�
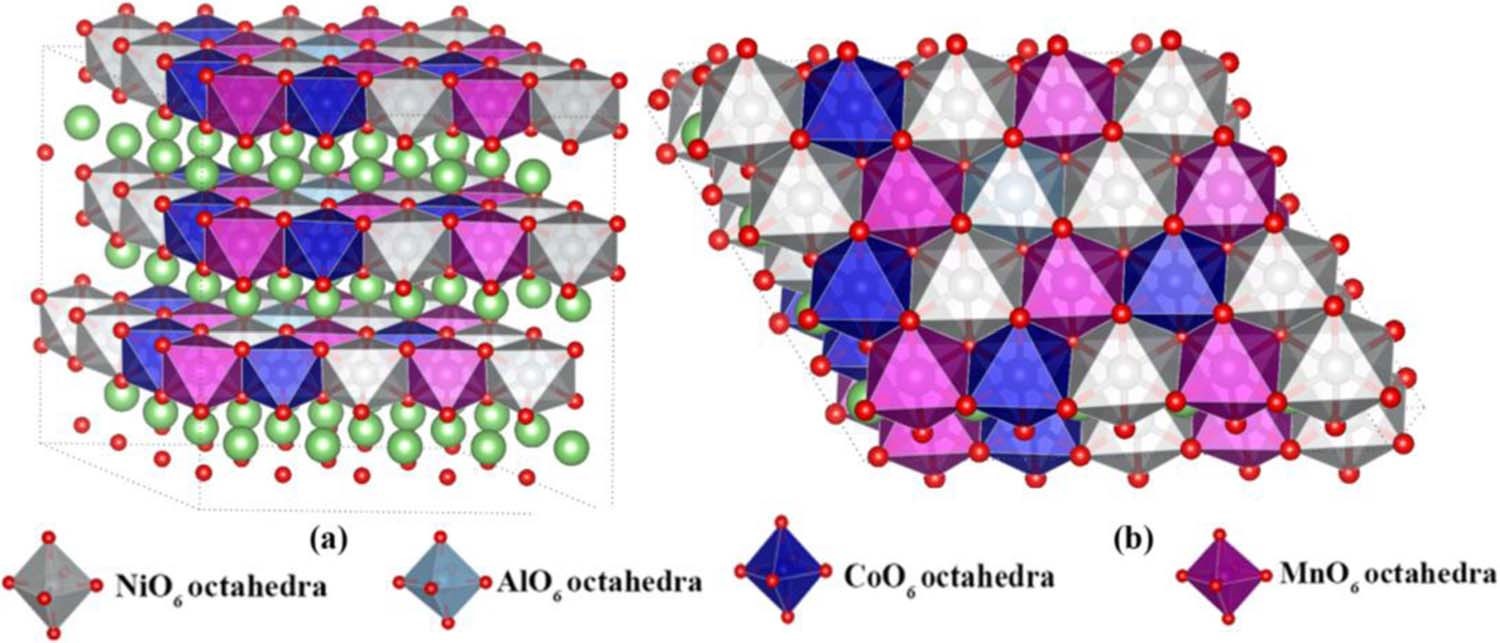
6. �ṹת��ģ���������缫��������ͽṹ�仯��˵���ṹ�ȶ��Ժ�ѭ���ԡ�
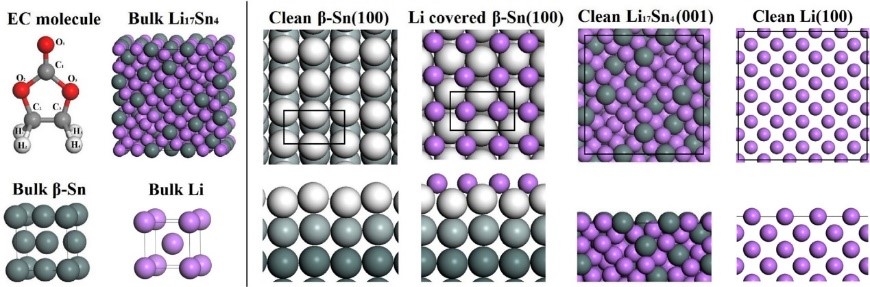
7. SEIĤ����ģ����SEIĤ���������ɷ֡��絼�ʡ���ѧ���ʵ�ģ�⡣
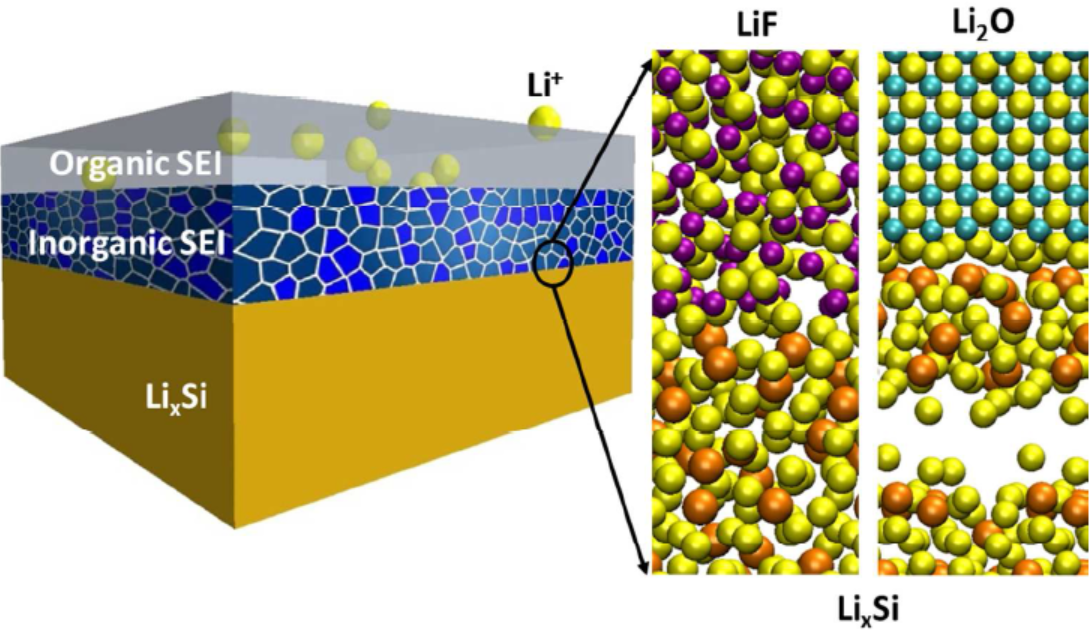
8. ��ѧ����ģ��������X�������վ�ϸ�ṹ��NEXAFS����NMR��ͼ��������ס�����ɼ����ס��������ȡ�
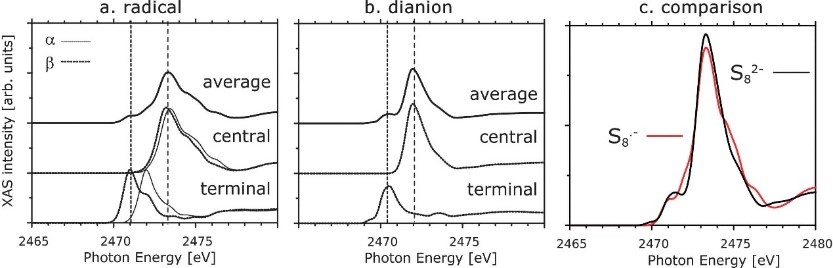
9. ��̬�����ģ���������̬����ʵ����ӵ絼�ʺ��ȶ��Եȡ�
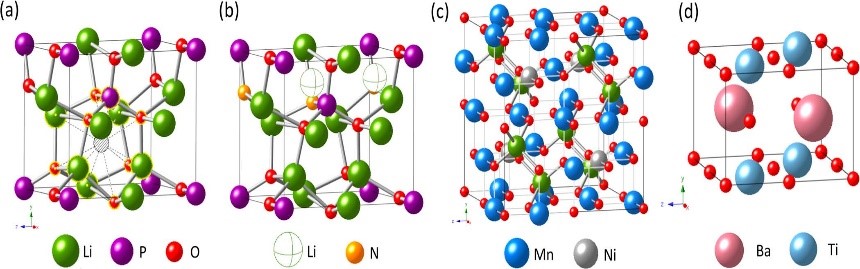
10.��ͼ����ģ�������������ͼԤ���м��ࡢ�о���ת�䡢�жϽṹ�ȶ��Եȡ�

��������ģ�����ܴ���̬�ܶȡ�ͶӰ�ֲ�̬�ܶȡ���ֵ���ܶȡ����Դ����������Խṹ�����ۼ������ӹ������ԭ��λ��ԭ�ӻ�ռλ�¶Ⱥ͵�ѹ�Ե缫���ϵ�Ӱ�졢�ܼ��������ܡ������ȶ��Ժ�����ѧ�ȶ��Եȡ�
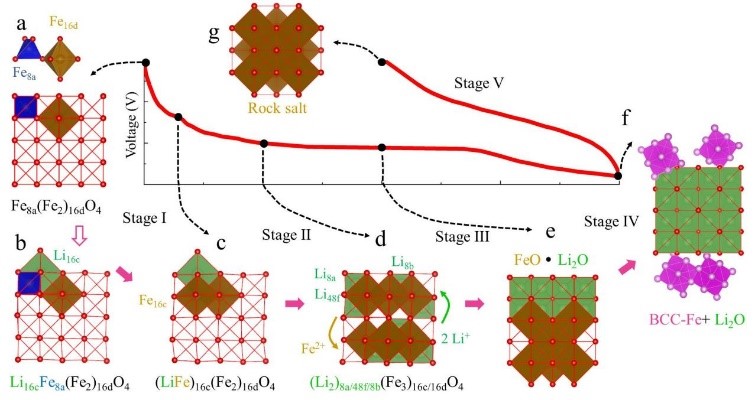
121����ز��ϼ��㰸�����붮����ģ���ڵ�������Ӧ��
����1����ԭ�Ӳ��ӵ�Эͬ�������ڵ�ԭ�Ӳ��ӣ�����-VASP��
�㽭��ҵ��ѧ������ͨ��DFT���㷢���������ӵ�̼���ܶԶ��������������Ҫǿ������������Լ�δ���ӵ�̼���ܣ��Ӷ�˵��������ԭ�Ӳ���֮�������ʮ��������ЭͬЧӦ��

Jin C, Zhang W, Zhuang Z, et al. Enhancedsulfide chemisorption using boron and oxygen dually doped multi-walled carbonnanotubes for advanced lithium�Csulfur batteries[J]. Journal of MaterialsChemistry A, 2017, 5(2): 632-640.
����2��Li������N����̼�����е���ɢ����ɢ-VASP��
��һ��ԭ������ķ����о�������е�����̼���ܣ����ֵ���������Ч�����ԭ������̼���ܺʹ���̼���ܵ����ݣ�ͬʱ������֮���γ�N-S�����Ӷ��Զ�������ǿ���������á�������һ���������Ԥ�����̼������һ�����õ�������������ϡ�
Wang Z, Niu X, Xiao J, et al. First principles prediction of nitrogen-doped carbon nanotubes as a high-performance cathode for Li�CS batteries[J]. Rsc Advances, 2013, 3(37): 16775-16780.
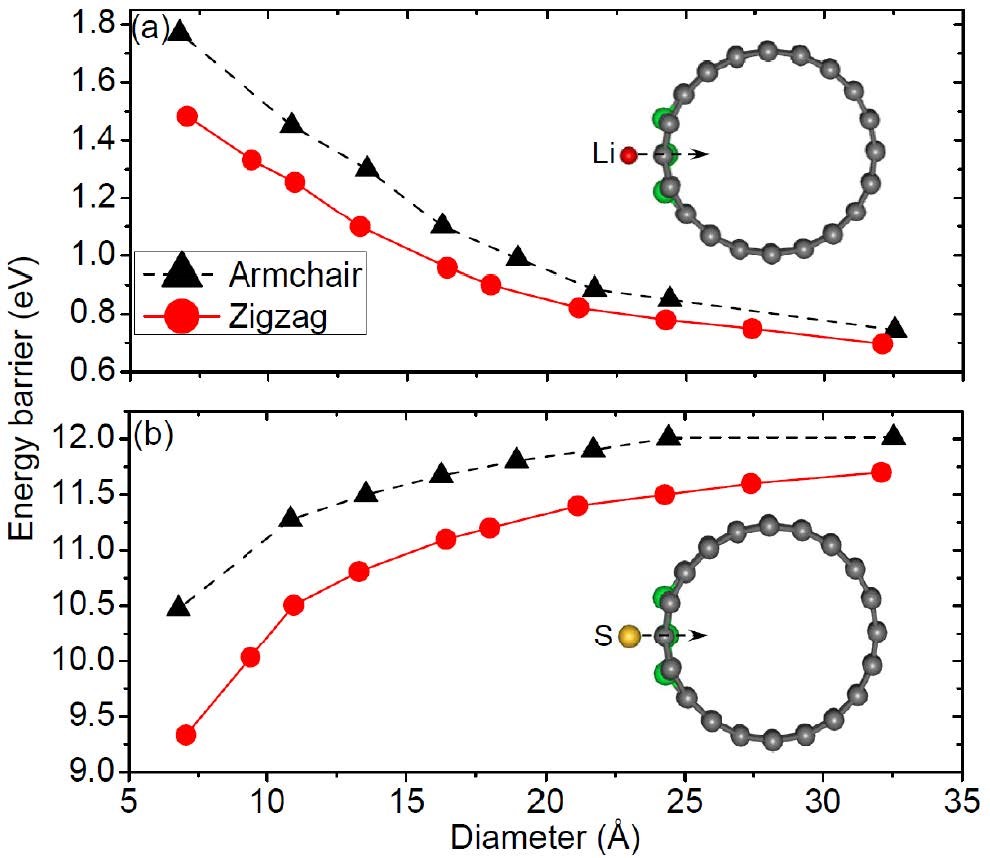
����3��ȱ�ݶ�����������Li����ɢ·��ģ�⣨����/��ɢ-VASP��
�п�λȱ�ݵ�ʯīϩ������LiS��LiS2��LiS3�������ʵ�Li��ɢ·�����������ߡ�
Liang Z, Fan X, Singh D J, et al. Adsorption anddiffusion of Li with S on pristine and defected graphene[J]. Physical Chemistry Chemical Physics, 2016, 18(45): 31268-31276.
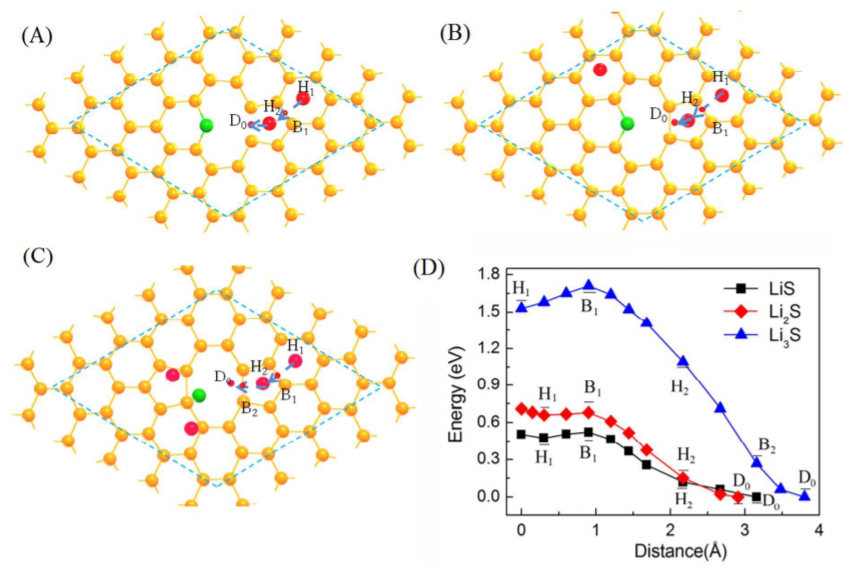
����4��ͨ����ͬ�м�̬�����ܵļ���Ԥ�ⷴӦ�������������������-VASP��
ͨ�������ܵõ��ŵ��м����Li2S�ڷŵ��ղ��� Li2Sx�������ʱ�Ļ�����Li2Sx��������Li2S��(110) �������(111)�������������������ֱ���γ�Li2S �������γ�Li2S2��
����ģ�����ijһ���̵������ܱ���Ϊ����ģ�������4������֮һ�����������Է����̵��оݣ�һ�����������ܽ��;����Է����С�
��111�����ϲ�ͬ��̬�ķ�����̬�Ͷ�Ӧ�ļ���˹�����ܣ����ԭʼ��G��
Liu Z, Hubble D, Balbuena P B, et al. Adsorption ofinsoluble polysulfides Li2S x (x= 1, 2) on Li2S surfaces[J].Physical Chemistry Chemical Physics, 2015, 17(14): 9032-9039.
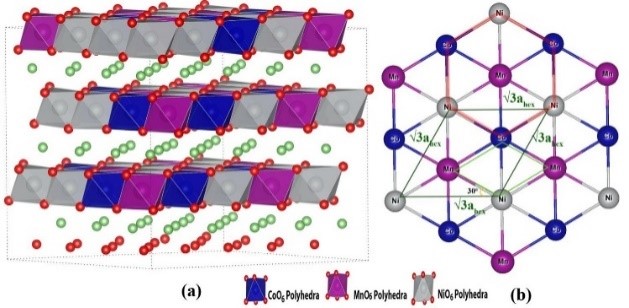
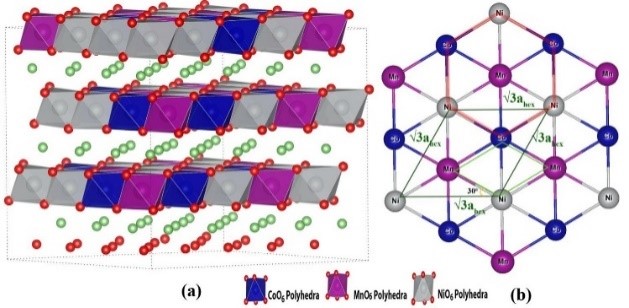
����5�����ӶԹ�̬�����LLZO���ӵ絼�ʺ��ȶ��Ե�Ӱ�죨�絼��/�ȶ���-VASP��
�Թ�̬����ʵ������о���Ҫ������������ӵ��ʺ��ȶ��ԣ���Ҳ����������ӵ�غ�����ع�̬����ʵĹ������⡣
�о���Rb��Ta����ԭ�Ӳ��ӶԹ�̬�����LLZO�����ӵ��ʺ��ȶ��Ե�Ӱ�죬 ������������ԭ�Ӳ��Ӳ����������ı��������LLZO�е�Ǩ��·�������������ı�LLZO������ӵ�Ũ�ȣ��Ӷ��ı�����ӵ��ʡ����⣬��������LLZO�����ߴ������ӵ���Ҳ����������Ӱ�죬�����ߴ��С����������ӵ��ʵ�Ѹ���½�����������LLZO�ľ����ߴ�ȴ���������Ե�Ӱ�졣
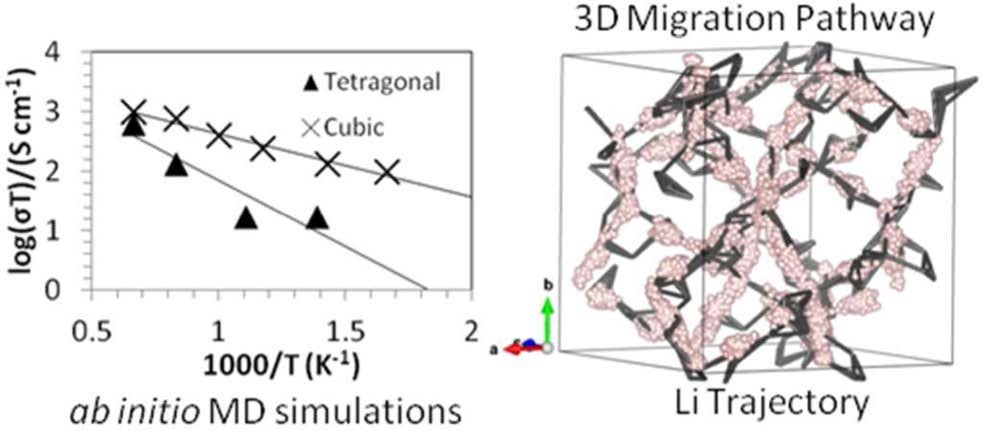
Miara L J, Ong S P, Mo Y, et al. Effect of Rb and Ta Doping on the Ionic Conductivity and Stability of the Garnet Li7+2x�Cy(La3�Cx Rbx)(Zr2�CyTay) O12(0�� x�� 0.375, 0�� y�� 1) Superionic Conductor: A First Principles Investigation[J]. Chemistry of Materials, 2013, 25(15): 3048-3055.
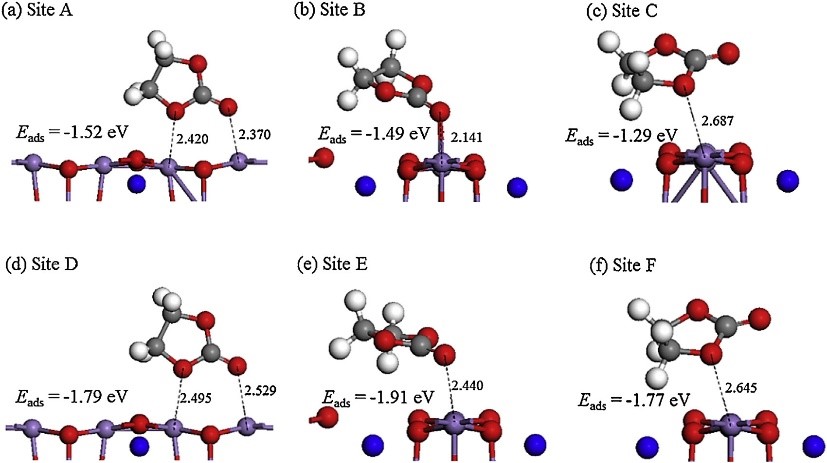
����6�����Һ��﮽������淴Ӧģ�⣨���淴Ӧ-VASP��
��AIMD��DFT���㷽���о���DME/LiTFSI��DOL/LiTFSI���Һ������֮����淴Ӧ������LiTFSI�����ڽ���ﮱ���ֽ�����LiF���ҷ�Ӧʮ��Ѹ�٣���DME��DOL�ܼ���ģ�����������ȶ���δ�����ֽⷴӦ��
Camacho-Forero L E, Smith T W, Bertolini S, et al.Reactivity at the lithium�Cmetal anode surface of lithium�Csulfur batteries[J].The Journal of Physical Chemistry C, 2015, 119(48): 26828-26839.
����7���о�����ӵ��������Ǩ���ʺ��ȶ��ԣ��絼��/�ȶ���-VASP��
ͨ����Ϸǵ�������ɢ��������ܶȷ��������о�������ӵ��������Ǩ���ʺ��ȶ��ԣ�ָ�������ӵ�����е͵����Ƶ�ʺ͵͵������̬�ܶȣ����͵�����������̬�ܶȻή�Ͳ��϶Ե绯ѧ�������ȶ��ԡ����ھ��е͵�﮴����ĺߵ������Ӵ����ģ����ʯ�ṹ���Ͼ��и����ӵ����Ժ��ȶ��ԣ�������Ϊ����ӵ��������ѡ��
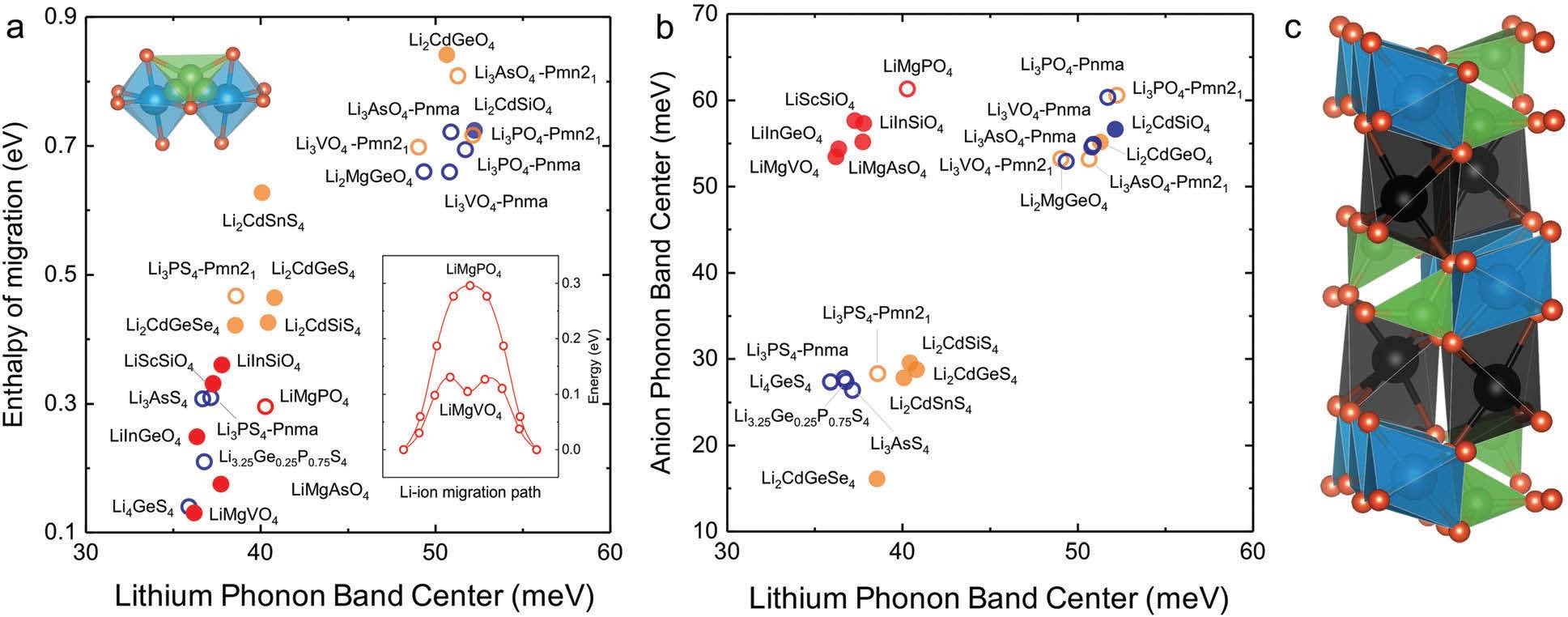
Muy S, Bachman J C,Giordano L, et al. Tuning mobility and stability of lithium ion conductors based on lattice dynamics[J]. Energy & Environmental Science, 2018, 11(4):850-859.
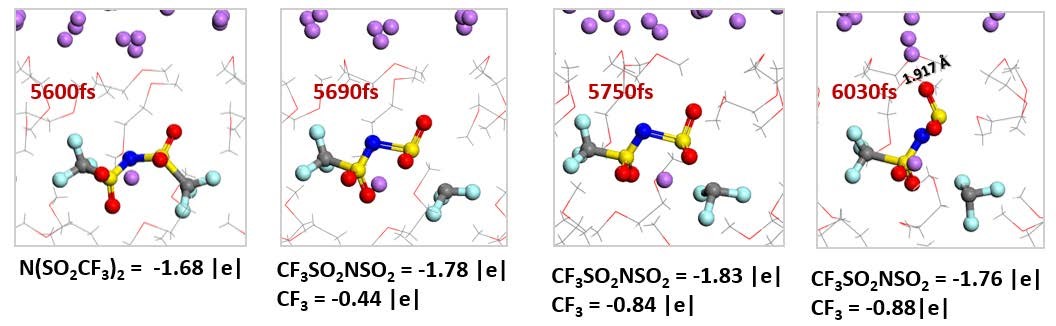
����8����һ��ԭ�������о�����Li�������֮��Ľ��淴Ӧѡ����(���淴Ӧ-VASP)
���Li-I2����е����Ӵ��������﮸����ķ�Ӧ��ͨ����һ��ԭ�������о��˽�����������֮��Ľ��淴Ӧѡ���ԡ����㷢�ֽ���﮵ģ�100���ͣ�110������Ե�����н�ǿ�ķ�Ӧ���ԣ���Ϊ�����γɵ⻯﮽���㡣�⻯﮵��ܽ�Ͱ�����Ϊ�����﮽���⻯﮵��γɺ�ȴ���ֱ�ӹ����ԣ�������⻯﮽Ϻ�ʱ���γɿ��ܽ�Ľ���㣬�Ӷ����Ƶ���Ӷ��ڽ���﮵Ľ�һ����ʴ���á�
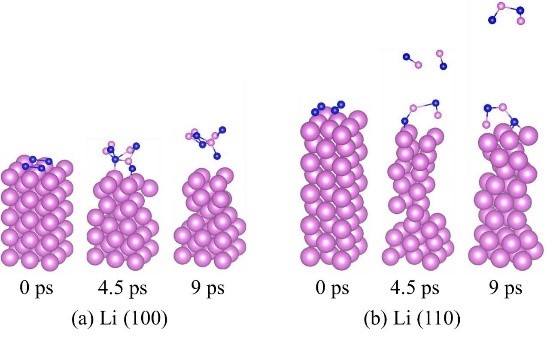
Liu Z, Hu W, Gao F, etal. An ab initio study for probing iodization reactions on metallic anodesurfaces of Li�CI2 batteries[J]. Journal of Materials Chemistry A, 2018, 6(17):7807-7814.
����9��Ceder��һ��ԭ���о���Ni�������ϵı�����ת�䣬�õ�Li-Ni-O����ͼ����ͼ-VASP��
ͨ����һ��ԭ�������о�����������������ѧ��ƽ�⣬���Դ��о���Ni�������ϵı�����ת�䡣�����˳����IJ�״���⾧ʯ����⾧ʯ��������ȶ���Li-Ni-Oͬ�����IJ��ϵ������������ڴ�Ԥ���м��࣬�õ�Li-Ni-O����ͼ��ͨ�����ؿ������ģ�ⲻͬ���XRDͼ����ͨ���Աȼ⾧ʯ����XRD ͼ������Ϊ�⾧ʯ�������ת�����Ϊ��-Ni1.75O2��Fm-3m����
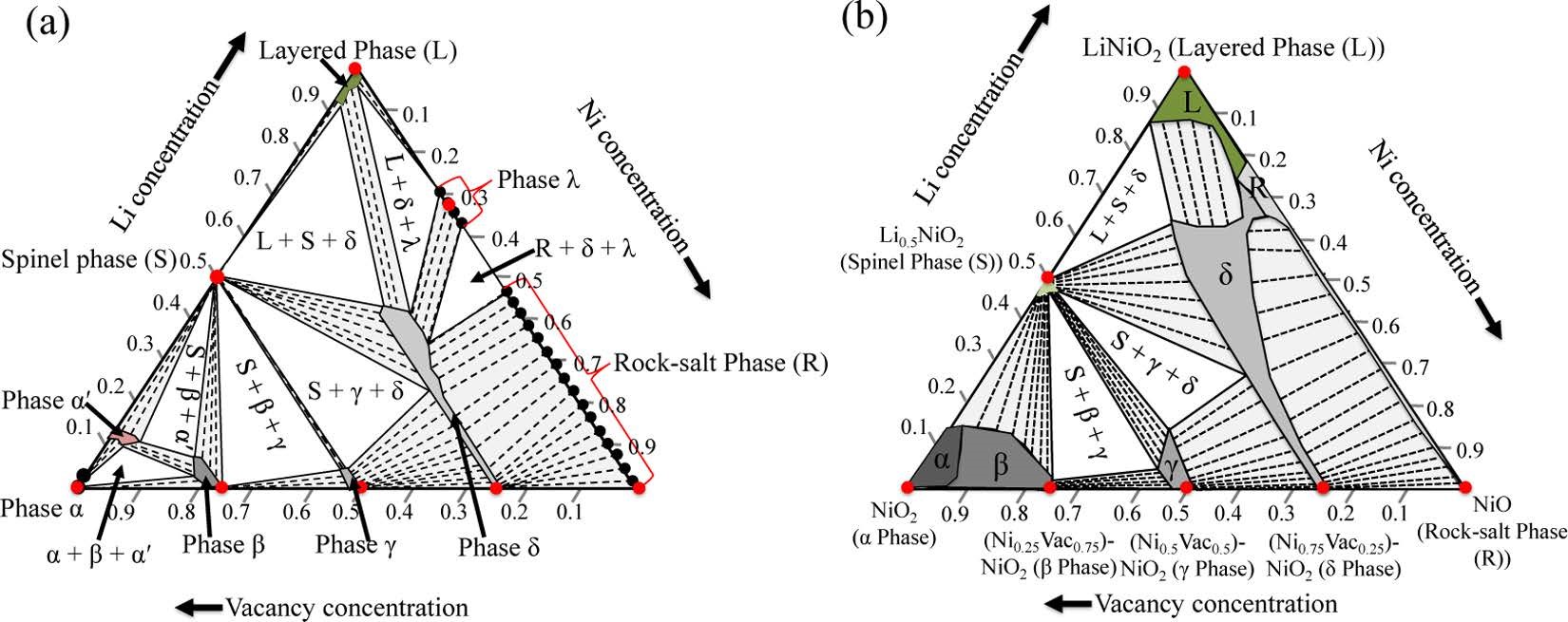
Das H, Urban A, Huang W, et al. First-Principles Simulation of the (Li�CNi�CVacancy) O Phase Diagram and Its Relevance for the Surface Phases in Ni-Rich Li-Ion Cathode Materials[J]. Chemistry of Materials,2017, 29(18): 7840-7851.
����10��������������ȥ﮻���Ӧ��Ԥ��������ݣ���ŵ����-VASP��
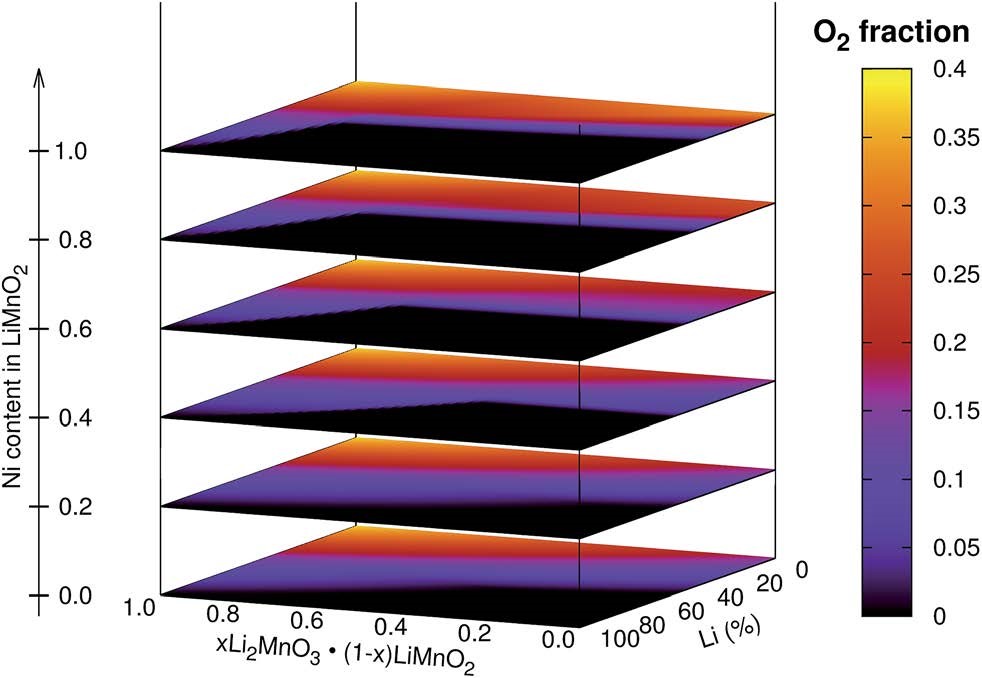
ͨ����һ��ԭ�������о����ڲ�ͬ�¶������£������-����������������ϵ�ȥ﮻���Ӧ�����ȶ��ԡ���ѧ�ƺͿ�·��ѹ��Ӱ�졣��״��������������ڳ������У���ҪΪ��⾧ʯ�ṹ�������ļ��뽵���˼⾧ʯ�ĺ�����ͨ��ȥ﮻������ж����ȶ��Եķ���������Ϊ���ټ⾧ʯ����γɺ�����������Li2MnO3.Li(Mn,Ni)O2�缫�����е�Li2MnO3��ֵĺ�����Ӧ����60%����LiMnO2����е�Ni����Ӧ��40%���ϡ�
Albina J M, Marusczyk A,Hammerschmidt T, et al. Finite-temperature property-maps of Li�CMn�CNi�CO cathode materials from ab initio calculations[J]. Journal of Materials Chemistry A,2018, 6(14): 5687-5694.
����11����һ��ԭ������Ԥ��BCx��Ϊ�������������ϵ�������֣�����-VASP��
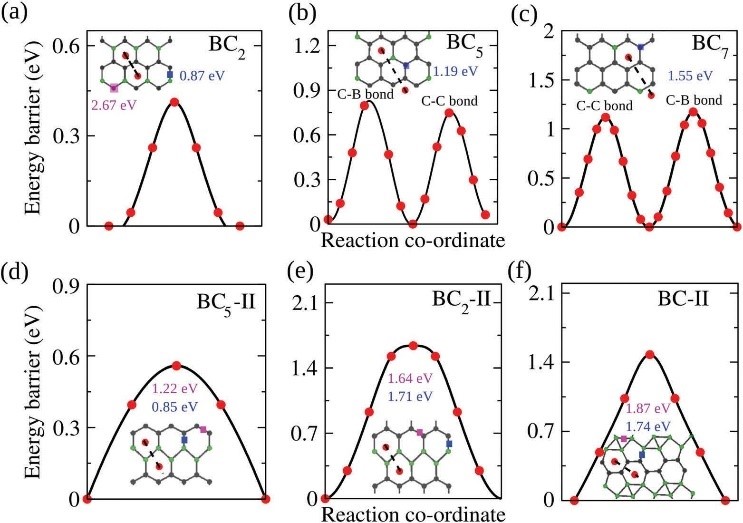
���õ�һ��ԭ�������о���B����ʯīϩBCx��Ϊ�������������ϵ����ԡ��������B���Ӻ�����ߴﵽx=2ʱ������������������������ߡ���Ϊ�������������BC2��������BCx���и���ķ����ܼ����ϵĿյ���̬��������BC2���������ɢ���ݺͿ�·��ѹ���������ͣ����и��õ�Li ����ѧ���ܡ�
Das D, Hardikar R P, HanS S, et al. Monolayer BC2: an ultrahigh capacity anode material forLi ion batteries[J]. Physical Chemistry Chemical Physics, 2017, 19(35):24230-24239.
��ԴSource��
http://www.cailiaoniu.com
http://www.nanoer.net
- ��һ� Science�еļ���ģ��
- ��һ� 1