 公司简介
公司简介
单原子催化
QQ学术交流群:1092348845
详细介绍
随着量子化学理论和分子模拟理论及高性能计算技术突飞猛进的发展,理论计算已经成为当代化学科学不可或缺的组成部分,也是催化科学实验研究的重要辅助手段。
DFT研究对了解单原子与载体之间相互作用和单原子催化剂优越的催化性能有不可替代的作用,主要用于以下两项分析:
确定最适单原子附着位/构型预测
实现方式:比较不同的单原子附着位点,比较键能、电子转移数、键长等参数及反应发生的实际条件。
选择最优反应路径/揭示反应机理。
实现方式:模拟反应气体的真实吸附、比较催化反应路径及速率控制步骤的能垒大小。
一、确定最适单原子附着位/构型预测
DFT 理论计算单原子可能附着的位点包括简单的表面吸附、取代载体空位或取代载体原子从而嵌入载体骨架
案例一
团队:福州大学化学化工学院林森等
研究:通过理论计算考察了不同单原子M( Ag,Au,Pt,Rh,Pd,Fe,Co 和Ir) 负载在具有B空缺的h-BN上的CO氧化机理
方法:从M-载体距离、M-N键长和键能、电子转移、金属与载体结合能、轨道杂化以及态密度,来说明单原子与B空缺型h-BN的几何构型和强相互作用。
结果:理论计算结果表明M-h-BN的金属位( Fe,Co,Ir 和Pd) 对O2的吸附较CO更稳定,且CO氧化反应服从E-R机理,其中Co-h-BN 在低温CO氧化反应中能垒最低。局域态密度分析金属的3d轨道与O2的2p轨道产生强杂化,使O2活化能垒显著降低。相比于其他金属位 ( Cu、Ag、Au、Pt 和Rh) ,CO吸附是强于O2,从而容易导致催化剂CO中毒。

案例二
团队:复旦大学化学系刘智攀团队
研究:对在ZrO2负载单原子Au催化1,3-丁二烯选择性氢化反应进行了DFT研究
方法:模拟4种不同的Au物种在四方二氧化锆载体的催化反应
结果:低温状态下Au(OH)3 在ZrO2上吸附最稳定,综合动力学模拟和Au(OH) 3 向Au(OH) 的转变,得出Au (OH)为最适宜的反应物种。Au (OH)以双配位的线性集合结构负载在ZrO2上,两个解离的H与Au-Olatt成键。反应过程中Au(OH)线性结构保持不变,Au离子与均相催化中Au配合物二者的几何结构类似,反应机理与均相催化中配体成键断键非常相似,说明均相催化和非均相催化之间的联系。

案例三
团队:埃因霍芬理工大学Hensen团队
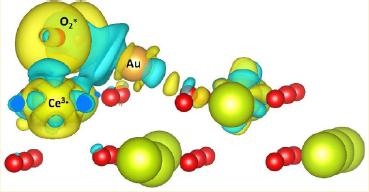
研究:考察了Au在CeO2 (110) 面的吸附、Au嵌入CeO2 ( 111) 面的氧空位
结果:与以前Au吸附在CeO2表面形成的对称结构的结论不同,发现更稳定的结构是O原子向Au方向偏移,与Ce和Au形成桥键,这种不对称结构能量更低更稳定。
Au嵌入CeO2 ( 111) 面的氧空位,导致Au-CeO2的电子重排,电子从Ce到转移到Au的6s轨道,形成Ce―Au共价键。此外研究人员还发现,CO的吸附能与Au的氧化态密切相关,Au具有更高价态,CO吸附更强。

案例四
团队:中国科学技术大学武晓君、张文华等
研究:考察单原子Au吸附在完美和具有空位的h-BN进行CO氧化反应
结果:发现单原子Au在完美的h-BN吸附能很低,极易解吸或发生团聚
考虑到B或N空位的存在可以为单原子提供稳定的附着位点,对Au原子在B和N空位的稳定性比较,利用分子动力学模拟O2、CO在B、N空位的吸附,发现在N空位O2的吸附能( 8. 81 eV) 远大于单原子Au(3. 17eV) 的吸附能,表明O2容易取代吸附在N空位Au原子。此外,实际产生B空位比N空位需要能量更低,所以DFT计算都是在单原子嵌入B空缺的情况下进行的。

相关学习文献:
[1] Lin S,Ye X,Johnson RS,Guo H. J. Phys. Chem. C,2013, 117( 33) : 17319.
[2] Liu Z P,Wang C M,FanK N. Angew. Chem. Int. Ed. , 2006,118( 41) : 7019.
[3] Song W,Hensen E J M. J. Phys. Chem. C,2013,117( 15) :7721.
[4] Mao K,Li L,Zhang W,Pei Y,Zeng X C,Wu X,Yang J. Sci. Rep.,2014,4. 5441.
二、反应机理分析
案例一
团队:美国波多黎哥大学陈中方团队
研究:受单原子催化剂Pt1 /FeOx的启发,考察了不同单原子( Au,Rh,Pd,Co,Cu,Ru 和 Ti) 分别负载到有氧空位和无氧空位的FeOx载体上对CO氧化反应的影响。
结果:单原子的存在能够降低氧空位的形成能垒,这与张涛等得出结论相一致。 有氧空位M1 /FeOx ( M = Rh,Pd,Co 和Cu) CO氧化反应遵从L-H机理,DFT计算结果见下图,反应分为三步,
第一步CO2的形成: OC* + O-O* →CO2 + O*,
第二步吸附的CO与化学吸附的O反应生成中间产物OCO* : CO* + O* →OCO* ,
第三步中间产物转变 成CO2 : OCO* →CO2。

对不同金属取代的研究显示,动力学和热力学结果表明Rh1/FeOx和Pd1 /FeOx催化剂比Pt1/FeOx有更好的CO催化氧化性能。
为了探讨催化剂对CO 氧化不同催化性能,他们分析催化剂的电子结构,对比不同M原子周围的投影态密度,发现M1/FeOx( M = Pt,Rh,Pd 和 Ru) 的d带中心明显向高能方向移动,因此催化剂具有更多的空d轨道从而CO与O2吸附分子具有更强的相互作用。他们又考察了O2的吸附,发现O2-p 轨道与M-d( M = Pt,Rh 和Pd) 轨道发生明显杂化,使O2更易活化便于CO氧化。
案例二
团队:橡树岭国家实验室Narula团队
研究:制备的单原子催化剂Pt /θ-Al2O3催化CO氧化、通过DRIFTS考察CO的吸附
结果:由于CO 和O2不能吸附在单原子Pt的临位,所以传统的L-H 机理不适用。考虑到有机金属配体进行均相催化和单原子Pt /θ-Al2O3(010) 与d10族配合物( Ph3P)2Pt 为等电子体,提出了优化后的L-H机理,反应机理如下图所示。

他们通过DRIFTS考察CO的吸附,不同Pt负载量催化剂样品在室温下形成了碳酸盐类中间产物,从而证实了CO氧化服从优化后的L-H 机理。DFT研究显示这两种氧化反应都符合改性的L-H 反应机理,即CO(或NO) 和O2共同吸附在Pt 单原子或中心上不与载体发生作用; 活性评价结果显示Pt /θ-Al2O3在两种反应中均具有良好的催化活性。
相关学习文献:
[1]靳永勇, 郝盼盼, 任军, & 李忠. (2015). 单原子催化――概念,方法与应用. 化学进展, 27(12), 1689-1704.
[2] Li F,Li Y,Zeng X C,Chen Z. ACS Catal. ,2014,5 ( 2) : 544.
[3] Moses-DeBusk M,Yoon M,Allard L F,Mullins D R,Wu Z, Yang X,Veith G,Stocks G M,Narula C K. J. Am. Chem. Soc. ,2013,135( 34): 12634.
DFT 计算的发展从电子原子尺度上使认识催化 剂的微观结构与反应机理成为可能,已经成为研究化学体系微观结构和化学反应机理的重要辅助手段,对于更好的理解单原子催化反应过程和反应机理,以及设计高效的单原子催化剂提供了可靠的理论支持。
- 上一款: Nature中理论计算
- 下一款: 1