
hotline��
17715390137
Tel/Wechat��
18101240246 (Technology)
0512-68565571
Email��mxenes@163.com ��Sales Engineer��bkxc.bonnie@gmail.com
Scan the code to follow or search the official account on WeChat:
2D Materials Fronrier After paying attention,
click on the lower right corner to contact us,
Enter enterprise WeChat.
Professional Services Online

Nearly 70% of the material-related work published by Science and Nature in the past three years has used computational simulation. At the same time, in the major top articles, especially when it comes to battery and catalytic mechanism analysis, theoretical simulation has become the main means of analysis and certification.
What is more embarrassing is that the gap between experiment and calculation is still very large. Many experimenters are not sure which problems can be solved by computational simulation, and how to apply it in the field of their own research!
We analyzed more than 5,000 applications of computational simulation in materials research, screened nearly a thousand high-quality cases, and finally selected 121 of the battery materials related work for readers.
It is worth noting that of the 121 calculation cases we randomly selected, 72 were calculated using the VASP software package. VASP is one of the most commonly used material computing commercial software. MedeA, as a VASP computing artifact, is a platform software that integrates the most advanced quantum mechanics VASP and molecular dynamics LAMMPS programs. It can be created under Windows system. Models, setup parameters, and visualization results are easy to use.
Battery material related calculation work,
mainly related to the following 10 directions
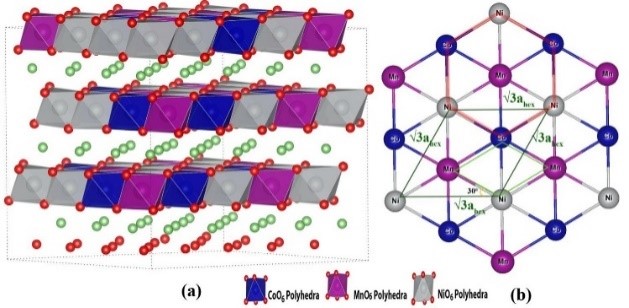
1. Adsorption calculation simulation: Calculate the adsorption energy, adsorption position, adsorption amount, etc. of a molecule in a specific material or crystal plane.
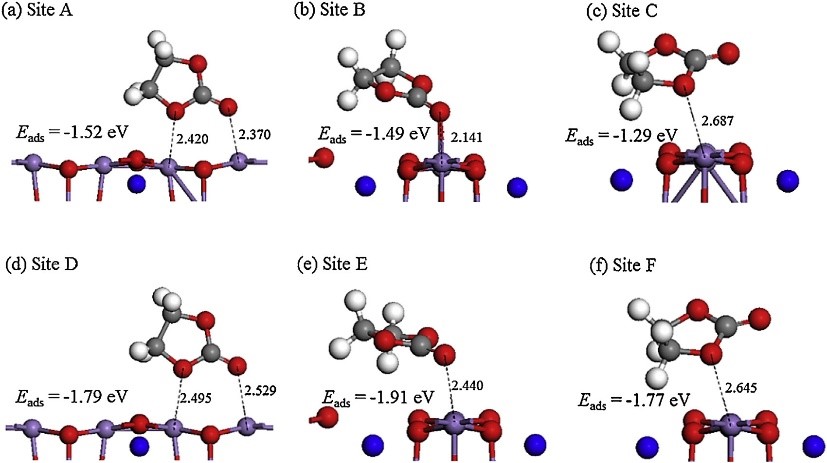
2. Reaction mechanism simulation: charge and discharge curve simulation at different current densities, phase transition during electrode material charge and discharge, reaction transition state simulation, capacity loss mechanism, reaction potential evolution, surface energy, charge transport mechanism, etc.
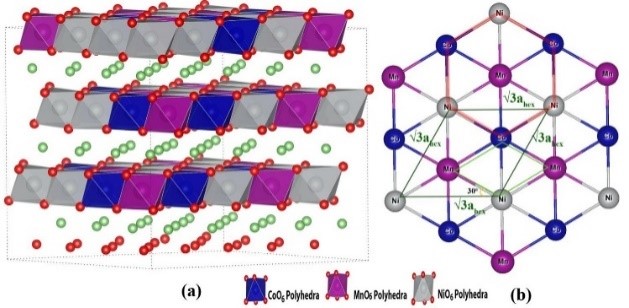
3. Ion diffusion simulation: Simulate the diffusion of ions in materials, calculate conductivity and diffusion migration paths.
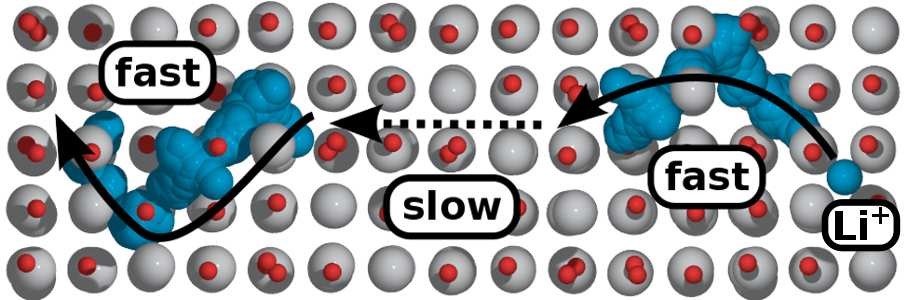
4. Material modification simulation: effects of doping/coating on electronic structure, lattice structure, phase structure, ionic conductivity, electronic conductivity, theoretical capacity, electrode potential, voltage curve, and open circuit voltage.
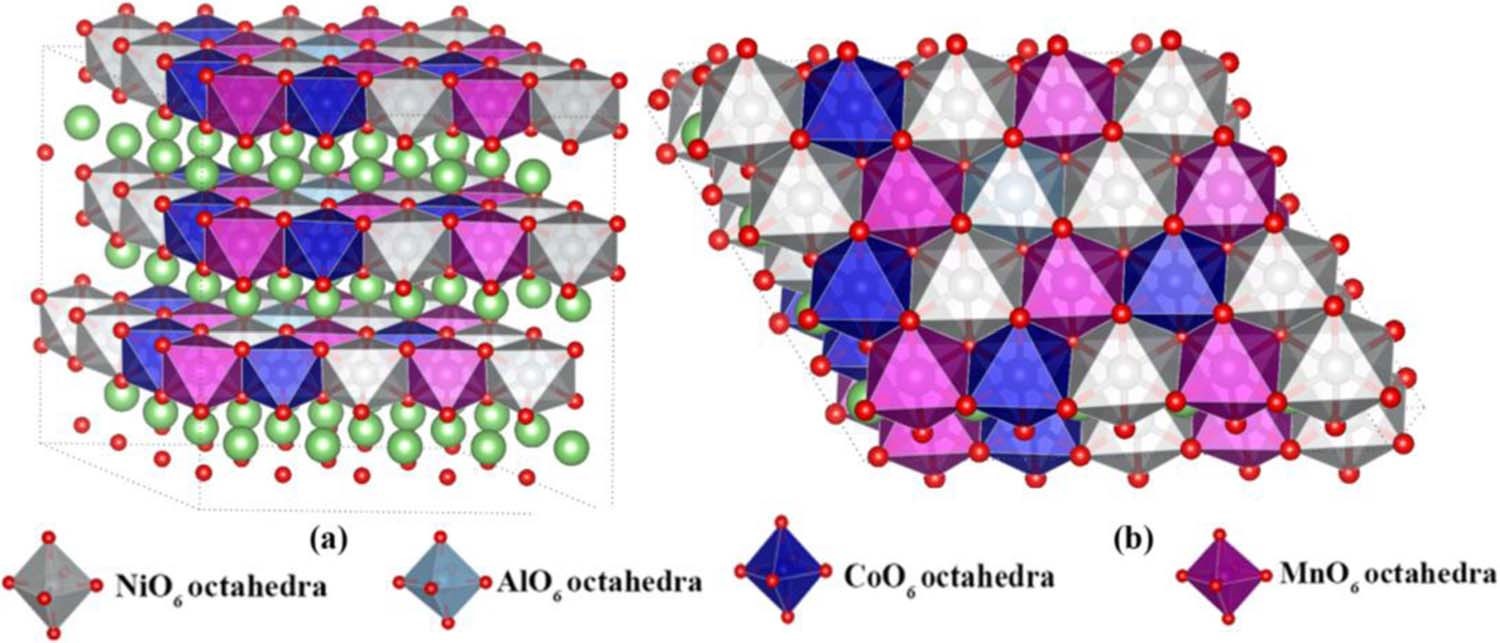
5. Interfacial reaction simulation: interaction between electrolyte/additive and electrode/SEI membrane interface, growth and inhibition mechanism of metallic lithium dendrites, reactivity of specific crystal faces, etc.
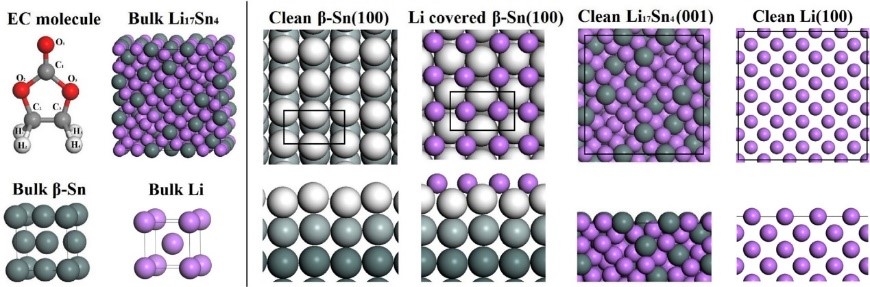
6. Structural transformation simulation: analysis of electrode material volume and structural changes, indicating structural stability and cycle.
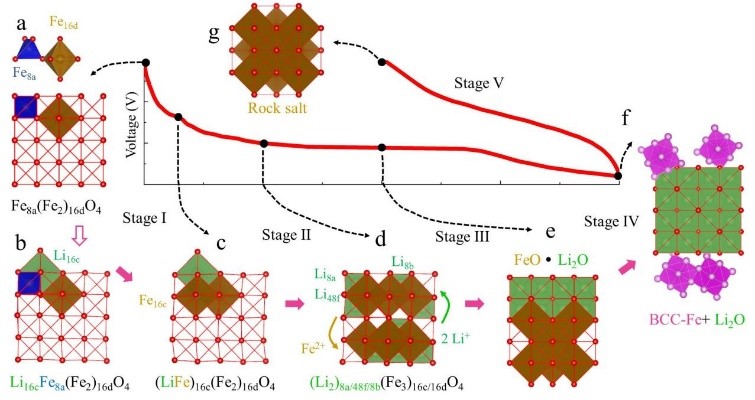
7. Simulation of SEI membrane mechanism: simulation of growth, composition, electrical conductivity and mechanical properties of SEI membrane.
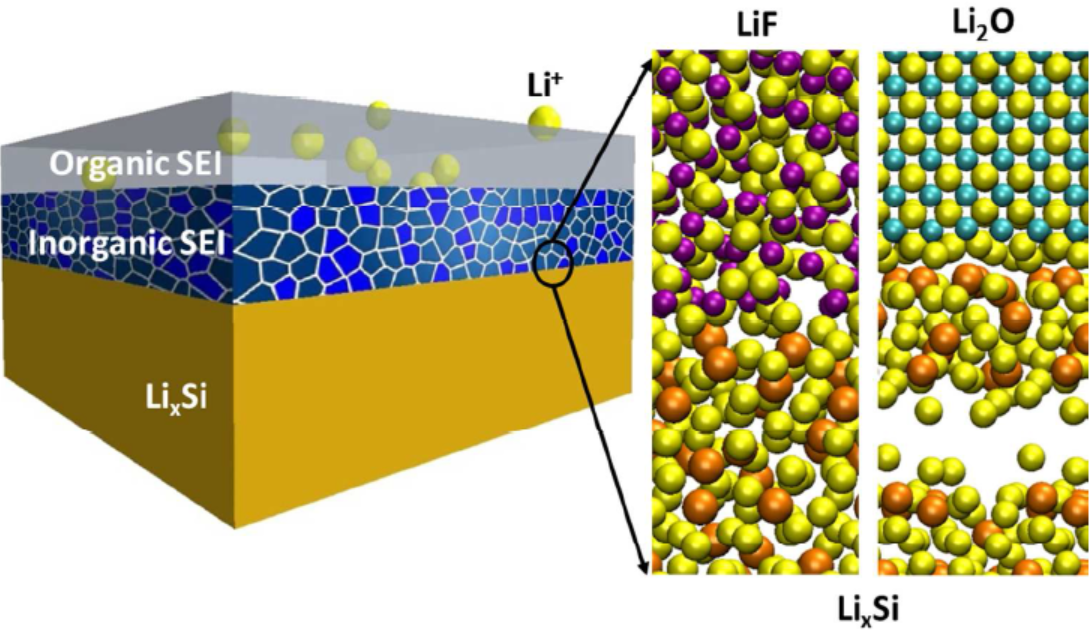
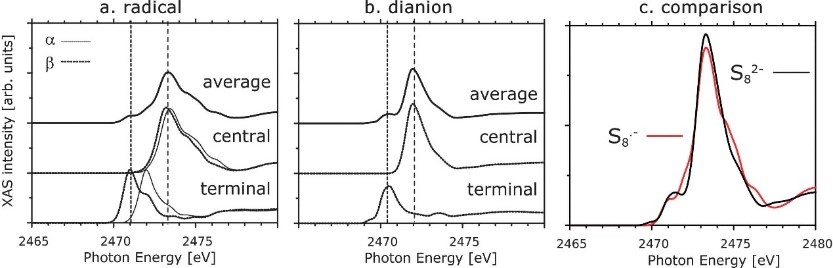
8. Spectral calculation simulation: near-edge X-ray absorption fine structure (NEXAFS), NMR spectrum, infrared spectrum, ultraviolet visible spectrum, Raman spectrum, etc.
9. Solid electrolyte simulation: Calculate the ionic conductivity and stability of solid electrolytes.
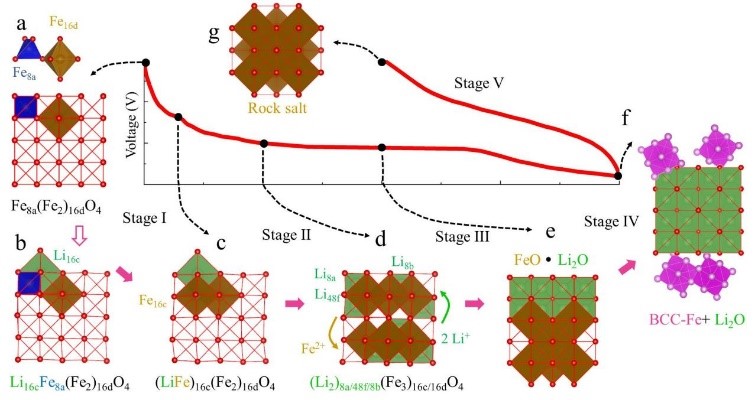
10. Phase diagram calculation simulation: Calculate the material phase diagram to predict the mesophase, study the phase transition, and judge the structural stability.

Other computational simulations: energy band, density of states, projected partial wave density, differential charge density, elastic band method, magnetic structure, covalent bond, molecular orbital, reduction potential, atomic mutual occupancy temperature, and voltage on electrode material, Solvation free energy, interface stability and thermodynamic stability.
121 battery material calculation cases, seconds to understand the application of simulation in the battery field
CASE1����ԭ�Ӳ��ӵ�Эͬ�������ڵ�ԭ�Ӳ��ӣ�����-VASP��

Jin C, Zhang W, Zhuang Z, et al. Enhancedsulfide chemisorption using boron and oxygen dually doped multi-walled carbonnanotubes for advanced lithium�Csulfur batteries[J]. Journal of MaterialsChemistry A, 2017, 5(2): 632-640.
CASE2��Li������N����̼�����е���ɢ����ɢ-VASP��
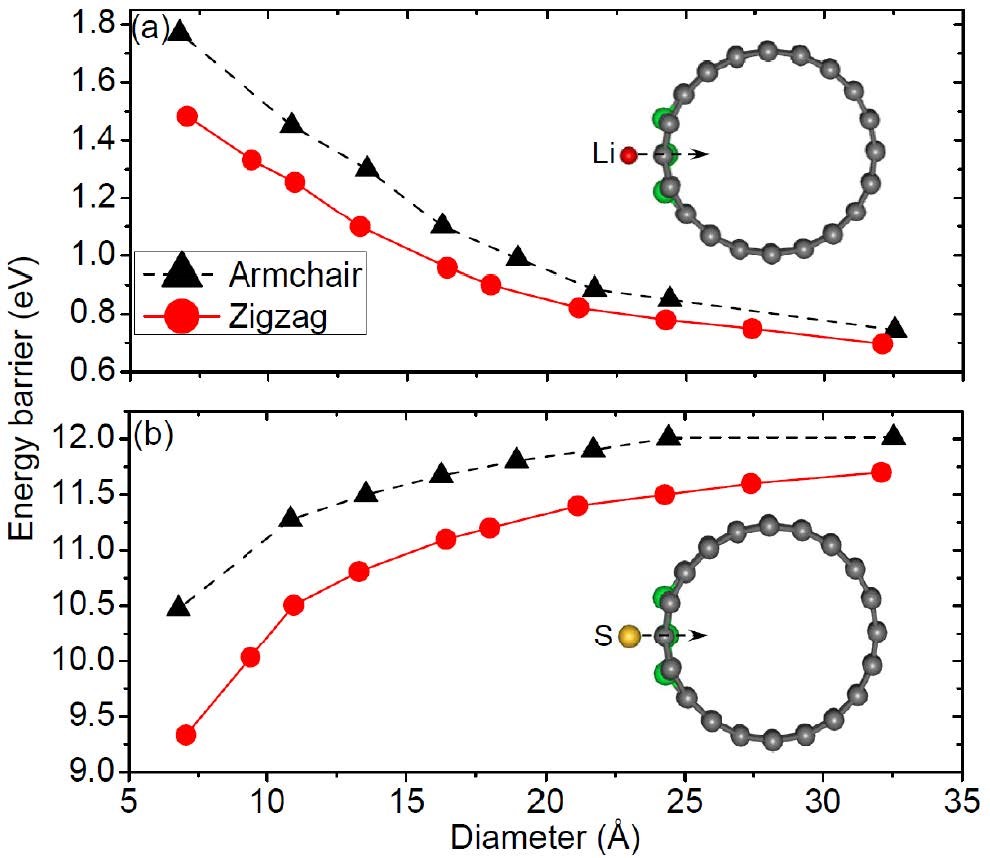
Wang Z, Niu X, Xiao J, et al. First principles prediction of nitrogen-doped carbon nanotubes as a high-performance cathode for Li�CS batteries[J]. Rsc Advances, 2013, 3(37): 16775-16780.
CASE3��ȱ�ݶ�����������Li����ɢ·��ģ�⣨����/��ɢ-VASP��
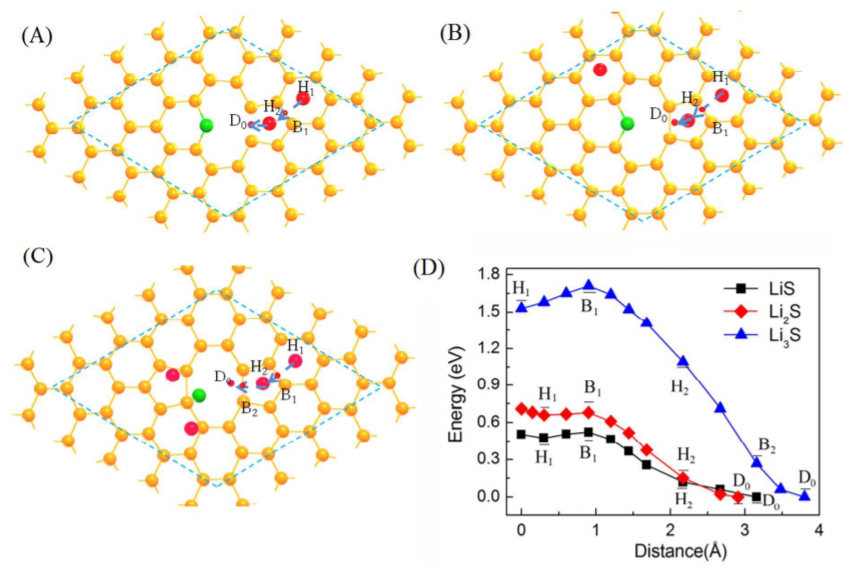
Liang Z, Fan X, Singh D J, et al. Adsorption anddiffusion of Li with S on pristine and defected graphene[J]. Physical Chemistry Chemical Physics, 2016, 18(45): 31268-31276.
CASE4��ͨ����ͬ�м�̬�����ܵļ���Ԥ�ⷴӦ�������������������-VASP��
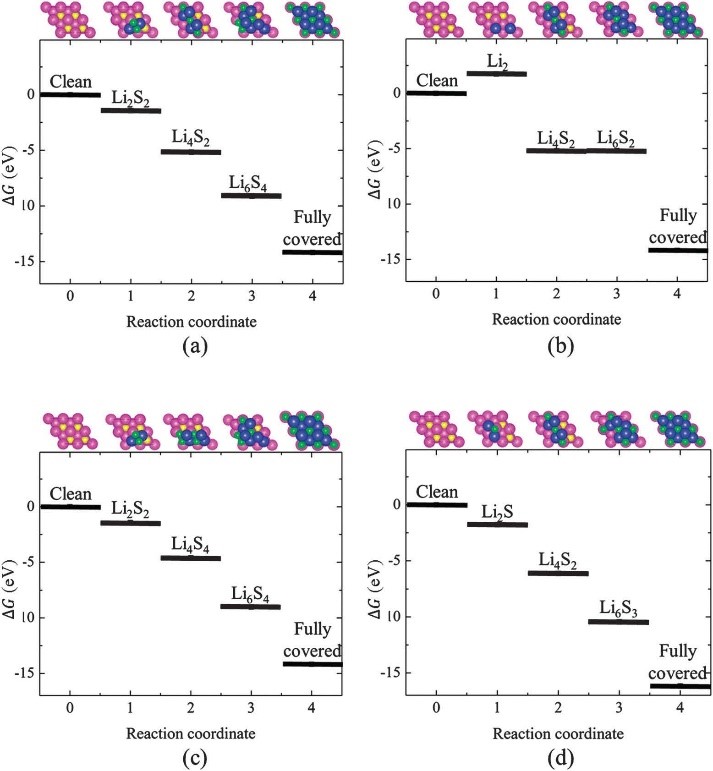
Liu Z, Hubble D, Balbuena P B, et al. Adsorption ofinsoluble polysulfides Li2S x (x= 1, 2) on Li2S surfaces[J].Physical Chemistry Chemical Physics, 2015, 17(14): 9032-9039.
CASE5�����ӶԹ�̬�����LLZO���ӵ絼�ʺ��ȶ��Ե�Ӱ�죨�絼��/�ȶ���-VASP��
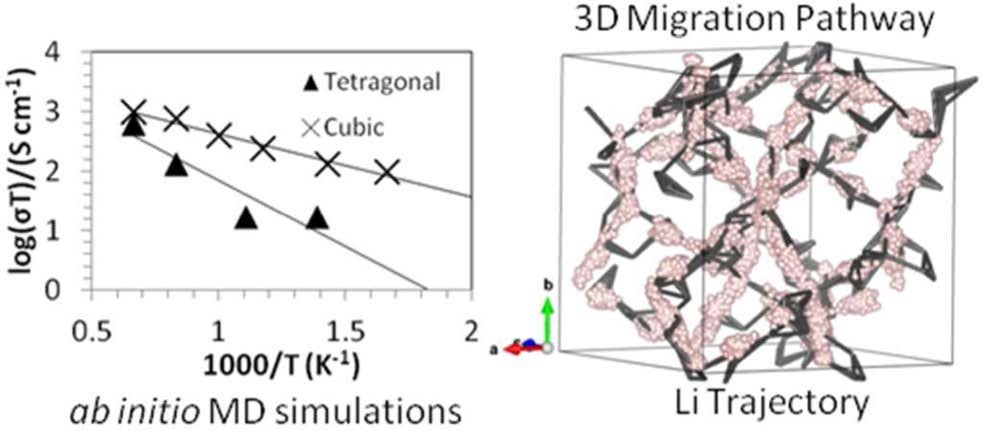
Miara L J, Ong S P, Mo Y, et al. Effect of Rb and Ta Doping on the Ionic Conductivity and Stability of the Garnet Li7+2x�Cy(La3�Cx Rbx)(Zr2�CyTay) O12(0�� x�� 0.375, 0�� y�� 1) Superionic Conductor: A First Principles Investigation[J]. Chemistry of Materials, 2013, 25(15): 3048-3055.
CASE6�����Һ��﮽������淴Ӧģ�⣨���淴Ӧ-VASP��
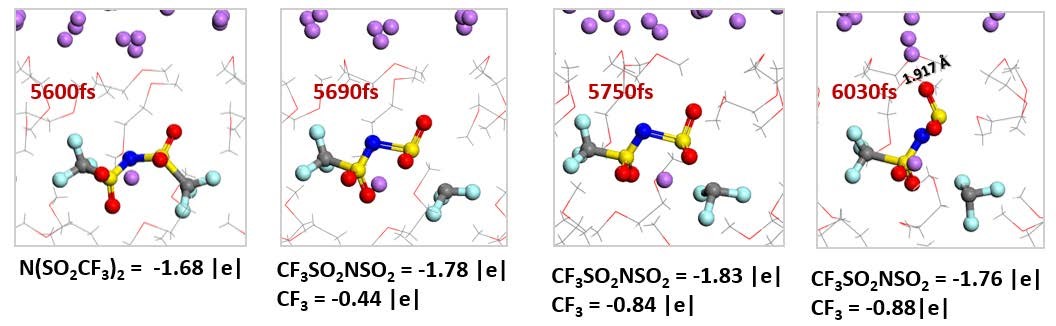
Camacho-Forero L E, Smith T W, Bertolini S, et al.Reactivity at the lithium�Cmetal anode surface of lithium�Csulfur batteries[J].The Journal of Physical Chemistry C, 2015, 119(48): 26828-26839.
CASE7���о�����ӵ��������Ǩ���ʺ��ȶ��ԣ��絼��/�ȶ���-VASP��
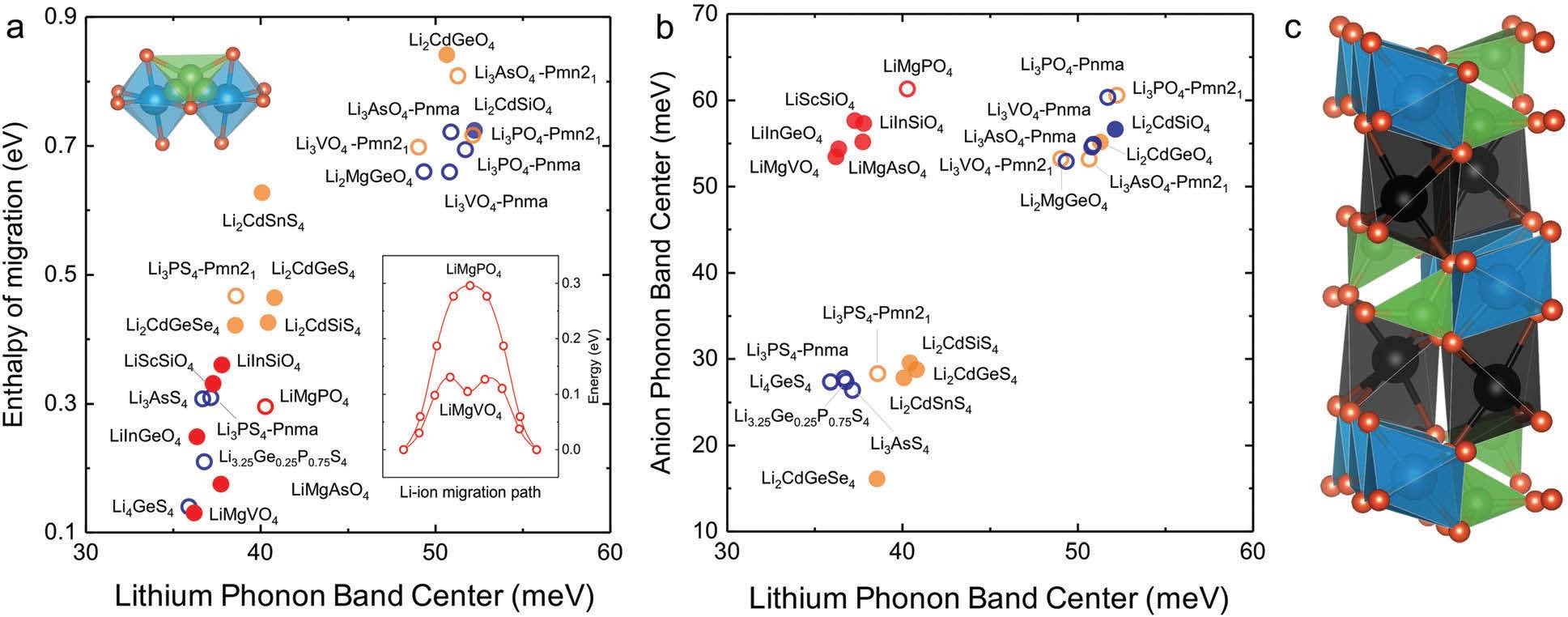
Muy S, Bachman J C,Giordano L, et al. Tuning mobility and stability of lithium ion conductors based on lattice dynamics[J]. Energy & Environmental Science, 2018, 11(4):850-859.
CASECASE8����һ��ԭ�������о�����Li�������֮��Ľ��淴Ӧѡ����(���淴Ӧ-VASP)
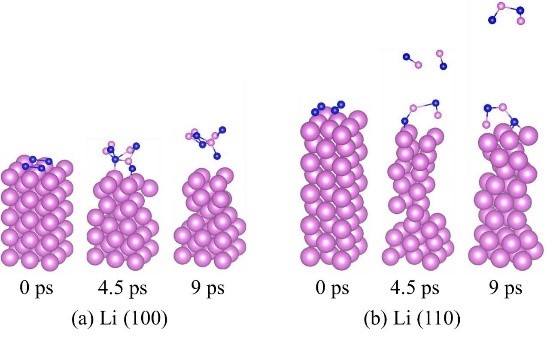
Liu Z, Hu W, Gao F, etal. An ab initio study for probing iodization reactions on metallic anodesurfaces of Li�CI2 batteries[J]. Journal of Materials Chemistry A, 2018, 6(17):7807-7814.
CASE9��Ceder��һ��ԭ���о���Ni�������ϵı�����ת�䣬�õ�Li-Ni-O����ͼ����ͼ-VASP��
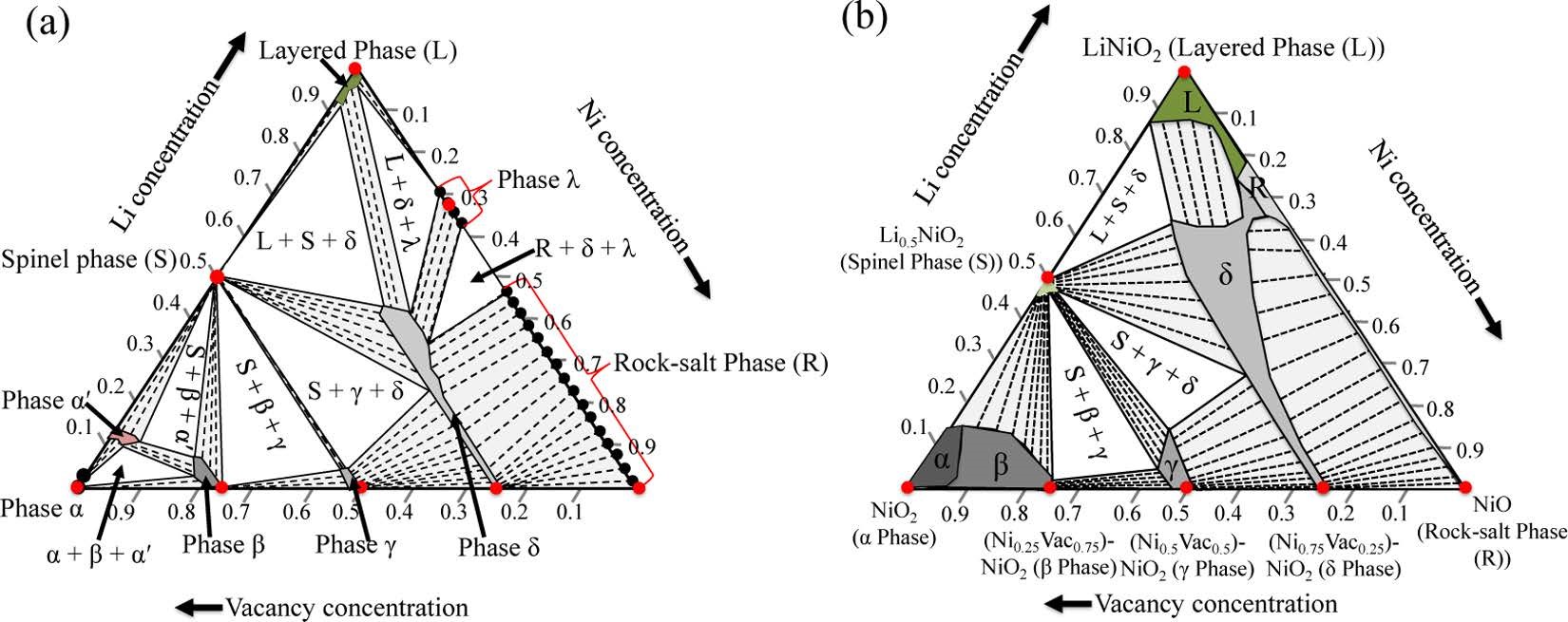
Das H, Urban A, Huang W, et al. First-Principles Simulation of the (Li�CNi�CVacancy) O Phase Diagram and Its Relevance for the Surface Phases in Ni-Rich Li-Ion Cathode Materials[J]. Chemistry of Materials,2017, 29(18): 7840-7851.
CASE10��������������ȥ﮻���Ӧ��Ԥ��������ݣ���ŵ����-VASP��
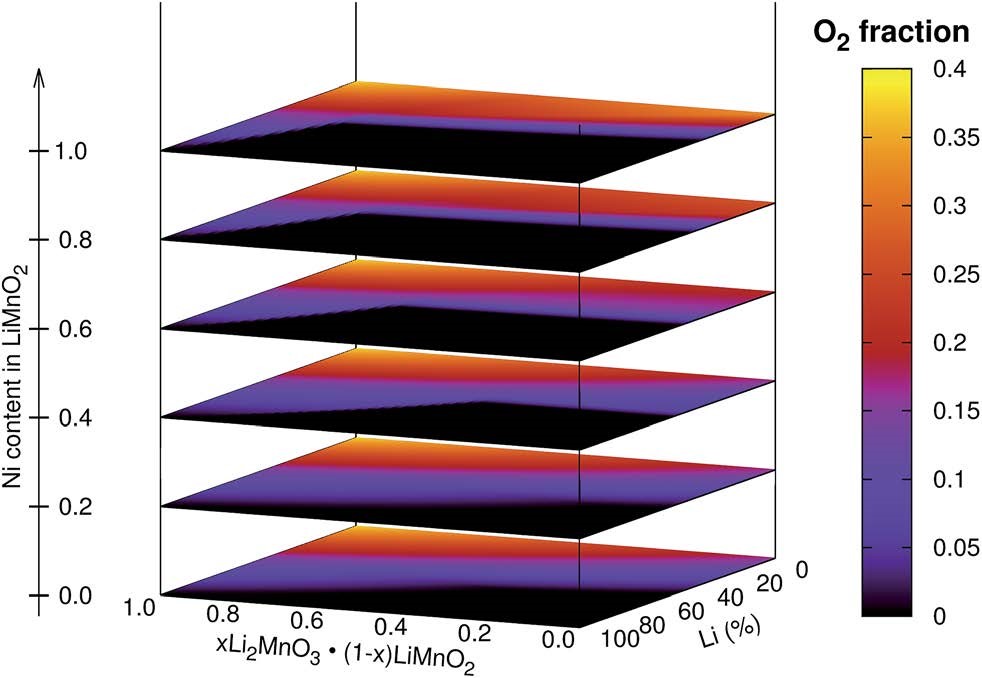
Albina J M, Marusczyk A,Hammerschmidt T, et al. Finite-temperature property-maps of Li�CMn�CNi�CO cathode materials from ab initio calculations[J]. Journal of Materials Chemistry A,2018, 6(14): 5687-5694.
CASE11����һ��ԭ������Ԥ��BCx��Ϊ�������������ϵ�������֣�����-VASP��
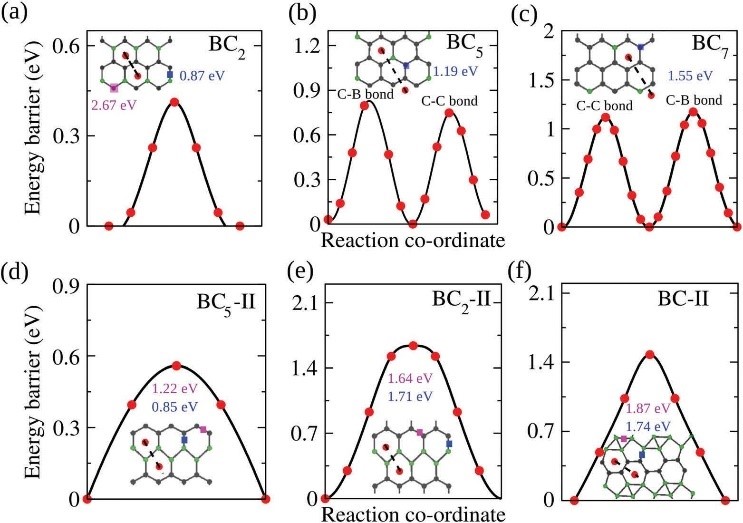
Das D, Hardikar R P, HanS S, et al. Monolayer BC2: an ultrahigh capacity anode material forLi ion batteries[J]. Physical Chemistry Chemical Physics, 2017, 19(35):24230-24239.
��ԴSource��
http://www.cailiaoniu.com
http://www.nanoer.net

| Reminder: Beijing Beike New Material Technology Co., Ltd. supplies products only for scientific research, not for humans |
| All rights reserved © 2019 beijing beike new material Technology Co., Ltd ��ICP��16054715-2�� |
