 公司简介
公司简介
VASP
QQ学术交流群:1092348845
详细介绍
密度泛函理论在理论化学领域已取得巨大的成功,能够提供详细的反应物在表面吸附的结构、吸附位、相互作用重构的详细信息,Kohn和Pople因在该领域做出了巨大贡献而获得1998年的诺贝尔化学奖。
DFT计算的发展使得从电子原子尺度上认识催化剂的微观结构与反应机理成为可能,已经成为研究化学体系微观结构和化学反应机理的重要辅助手段,对于更好的理解催化反应过程和反应机理,以及设计高效的催化剂提供了可靠的理论支持。
在材料计算领域,VASP是目前材料计算使用的最普遍的商用软件之一,在以下11个案例中,使用VASP计算的有8个。它全称为“Vienna Ab-initio Simulation Package”,由维也纳大学的Hafner小组开发的。VASP能进行电子结构和分子动力学模拟,可以计算材料的几何性质(键长、键角、晶格常数),电子结构性质(电子态密度、能带结构、电子密度分布、电子局域函数),光学性质(介电常数、吸收光谱、折射率)以及磁学性质等。
VASP功能强大但短板之处在于它没有一个很好的操作界面,MedeA成功解决了这个问题,它是一款整合了最先进的量子力学VASP和分子动力学LAMMPS程序的一个平台软件,可以实现在Windows系统下创建模型、设置参数及可视化结果分析,易于操作使用,MedeA功能强大且没有盗版资源。
1.Nature Nanotechnology:DFT计算单原子催化剂中不同Pt原子的反应路径,研究单原子相互作用
中科大研究了关于单原子催化中的单原子相互作用,研究人员利用DFT量化计算,揭示了催化CO2加氢过程中相邻Pt单原子间的协同作用。
计算模拟结果表明,孤立Pt单原子路径在CO2加氢制甲醇过程中不经历甲酸中间物种,限速步在CH2OH*-CH3OH,因此CH2OH*为主要中间物种。而近邻Pt单原子路径在CO2加氢制甲醇过程中有甲酸中间物种,限速步在COOH*-HCOOH,因此COOH*为主要中间物种。
孤立Pt单原子路径与近邻Pt单原子路径的不同,从而揭示两种单原子在催化中完全不同的催化性能,佐证近邻Pt单原子之间存在明显协同作用。
Li H, Wang L, Dai Y, etal. Synergetic interaction between neighbouring platinum monomers in CO2 hydrogenation[J]. Nature nanotechnology, 2018, 13(5): 411.
2. 使用VASP研究催化活性位点,阐明催化剂高活性的内在原因
天津大学邓意达、胡文彬教授团队提出了一种简便、快速可控合成氮掺杂石墨烯负载不同晶面包裹Co3O4纳米结构的新方法,调控Co3O4表面Co2+和Co3+活性位点的比例并研究表面氧的吸、脱附行为。该催化剂表现出优异的氧还原和析氧的双功能活性,并能增强金属-空气电池性能。
文章也是采用计算模拟(VASP软件包)的强大功能,利用密度泛函理论(DFT)阐述Co3O4表面八面体间隙的三价Co活性位,同时揭示Co3O4与载体氮掺杂石墨烯载体之间的耦合协同作用,阐明催化剂高活性的内在原因。
Han X,He G, He Y, et al. Engineering Catalytic Active Sites on Cobalt Oxide Surfacefor Enhanced Oxygen Electrocatalysis[J]. Advanced Energy Materials, 2018,8(10): 1702222.
3.构建Mo2N-Mo2C体系,使用VASP计算吸附位点和自由能确定催化活性中心
黑龙江大学付宏刚教授(通讯作者)科研团队,通过原位构建多孔还原氧化石墨烯负载的氮、碳化钼异质结用于高效的电催化析氢反应(HER)。
仍是使用VASP量化软件包,基于密度泛函理论,计算了在Mo2N-Mo2C体系上可能的吸附位点以及基于各种研究系统计算的HER自由能,计算指出Mo2N-Mo2C界面处的N-Mo-C是主要的催化活性中心,多孔还原氧化石墨烯作为载体主要起到传质以及电荷传递的作用。
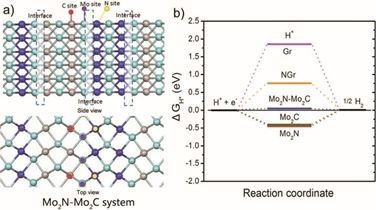
Yan,Haijing, et al. "Holeyreduced graphene oxide coupled with an Mo2N–Mo2Cheterojunction for efficienthydrogen evolution." Advanced Materials 30.2(2018)
4. 使用VASP软件包计算研究不同单原子的CO氧化机理
福州大学化学化工学院林森课题组通过理论计算考察了不同单原子M( Ag,Au,Pt,Rh,Pd,Fe,Co 和Ir) 负载在具有B空缺的h-BN上的CO氧化机理。 从M-载体距离、M-N键长和键能、电子转移、金属与载体结合能、轨道杂化以及态密度,来说明单原子与B空缺型h-BN的几何构型和强相互作用。
理论计算结果表明M-h-BN的金属位( Fe,Co,Ir 和Pd) 对O2的吸附较CO更稳定,且CO氧化反应服从E-R机理,其中Co-h-BN 在低温CO氧化反应中能垒最低。局域态密度分析金属的3d轨道与O2的2p轨道产生强杂化,使O2活化能垒显著降低。相比于其他金属位 ( Cu、Ag、Au、Pt 和Rh) ,CO吸附是强于O2,从而容易导致催化剂CO中毒。
Lin S, Ye X, Johnson RS, et al. First-principles investigations of metal (Cu, Ag, Au, Pt, Rh, Pd, Fe,Co, and Ir) doped hexagonal boron nitride nanosheets: stability and catalysisof CO oxidation[J]. The Journal of Physical Chemistry C, 2013, 117(33): 17319-17326.
5. 使用VASP研究单原子表面吸附,确定最稳定结构
埃因霍芬理工大学Hensen团队使用VASP软件包进行DFT计算模拟,考察了Au在CeO2 (110) 面的吸附、Au嵌入CeO2 ( 111) 面的氧空位。与以前Au吸附在CeO2表面形成的对称结构的结论不同,发现更稳定的结构是O原子向Au方向偏移,与Ce和Au形成桥键,这种不对称结构能量更低更稳定。
Au嵌入CeO2 ( 111) 面的氧空位,导致Au-CeO2的电子重排,电子从Ce到转移到Au的6s轨道,形成Ce—Au共价键。此外研究人员还发现,CO的吸附能与Au的氧化态密切相关,Au具有更高价态,CO吸附更强。
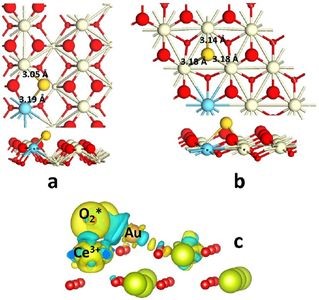
Song W, Hensen E J M.Structure sensitivity in CO oxidation by a single Au atom supported onceria[J]. The Journal of Physical Chemistry C, 2013, 117(15): 7721-7726.
6. 利用VAS软件包使用DFT方法分析单原子催化的反应机理
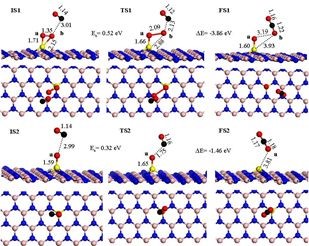
美国波多黎哥大学陈中方团队受单原子催化剂Pt1 /FeOx的启发,考察了不同单原子(Au,Rh,Pd,Co,Cu,Ru 和 Ti) 分别负载到有氧空位和无氧空位的FeOx载体上对CO氧化反应的影响。
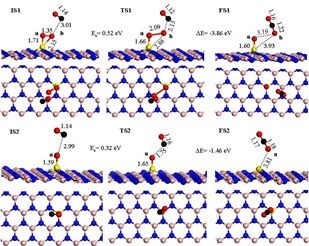
单原子的存在能够降低氧空位的形成能垒,这与张涛等得出结论相一致。有氧空位M1/FeOx ( M = Rh,Pd,Co 和Cu) CO氧化反应遵从L-H机理,DFT计算结果见下图,反应分为三步。
第一步CO2的形成: OC* + O-O* →CO2 + O*,
第二步吸附的CO与化学吸附的O反应生成中间产物OCO* : CO* + O* →OCO* ,
第三步中间产物转变成CO2: OCO* →CO2。
对不同金属取代的研究显示,动力学和热力学结果表明Rh1/FeOx和Pd1/FeOx催化剂比Pt1/FeOx有更好的CO催化氧化性能。
为了探讨催化剂对CO 氧化不同催化性能,他们分析催化剂的电子结构,对比不同M原子周围的投影态密度,发现M1 /FeOx(M = Pt,Rh,Pd 和 Ru) 的d带中心明显向高能方向移动,因此催化剂具有更多的空d轨道从而CO与O2吸附分子具有更强的相互作用。他们又考察了O2的吸附,发现O2-p 轨道与M-d( M = Pt,Rh 和Pd) 轨道发生明显杂化,使O2更易活化便于CO氧化。
Li F, Li Y, Zeng X C, etal. Exploration of high-performance single-atom catalysts on support M1/FeO xfor CO oxidation via computational study[J]. ACS Catalysis, 2014, 5(2):544-552.
7. 实验+DFT模拟探究Pt /θ-Al2O3催化CO氧化反应机理
美国橡树岭国家实验室Narula团队制备的单原子催化剂Pt /θ-Al2O3催化CO氧化、通过DRIFTS考察CO的吸附,使用VASP软件包进行DFT计算模拟。
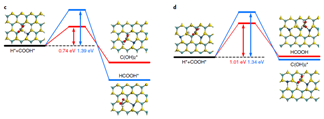
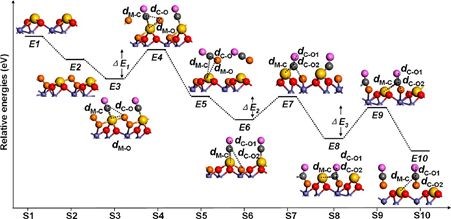
由于CO 和O2不能吸附在单原子Pt的临位,所以传统的L-H 机理不适用。考虑到有机金属配体进行均相催化和单原子Pt /θ-Al2O3(010)与d10族配合物( Ph3P)2Pt为等电子体,提出了优化后的L-H机理,反应机理如下图所示。
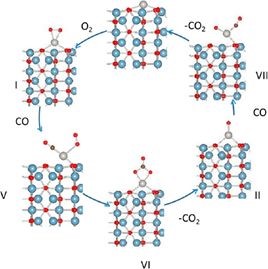
他们通过DRIFTS考察CO的吸附,不同Pt负载量催化剂样品在室温下形成了碳酸盐类中间产物,从而证实了CO氧化服从优化后的L-H 机理。DFT研究显示这两种氧化反应都符合改性的L-H 反应机理,即CO(或NO) 和O2共同吸附在Pt 单原子或中心上不与载体发生作用; 活性评价结果显示Pt /θ-Al2O3在两种反应中均具有良好的催化活性。
8. 利用VASP软件包进行充分DFT模拟后精准合成Pd-Cu双金属合金催化CO2加氢制甲醇
大连理工大学聂小娃副教授采用密度泛函理论(DFT)计算结合实验研究的方法,对Pd-Cu双金属合金催化CO2加氢合成甲醇的反应机理进行了深入研究,揭示了双金属合金结构及水对CO2转化及甲醇选择性的重要影响。
DFT计算研究结果表明,阶梯型PdCu(111)表面具有低配位的Pd原子暴露在表面上,其对CO2和H2的吸附活化比表面富含Cu的平面型PdCu3(111)合金具有更高的活性,同时其对CO2的初始加氢转化也表现出更优异的催化性能。DFT计算预测Pd-Cu型合金是CO2加氢制甲醇的优良催化剂。
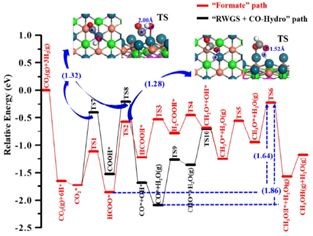
通过计算反应路径和基元反应能量学,提出了PdCu(111)表面上甲醇生成的优势反应路径:
CO2*→HCOO*→HCOOH*→H2COOH*→CH2O*→CH3O*→CH3OH*
基于DFT计算结果,美国宾夕法尼亚州立大学能源研究所的姜潇博士后进行了系统的实验研究,合成了富含PdCu(111)合金相及富含PdCu3(111)合金相的双金属催化剂,与理论计算模型相一致。
Nie X, Jiang X, Wang H, et al.Mechanistic Understanding of Alloy Effect and Water Promotion for Pd-CuBimetallic Catalysts in CO2 Hydrogenation to Methanol[J]. ACS Catalysis, 2018,8(6): 4873-4892.
9. 利用VASP结合过渡态搜索和AIMD方法成功解释实验现象,揭示动力学在多相催化反应中的重要性
福州大学化学化工学院林森课题组首先利用DFT计算来阐明SAC活性位点的性质和1,3-丁二烯加氢反应催化机理。在第二加氢过程中,后过渡态动力学驱动目标产物1-丁烯的脱附,而不是进一步加氢生成丁烷,从而成功地解释了实验观察到的高选择性现象。
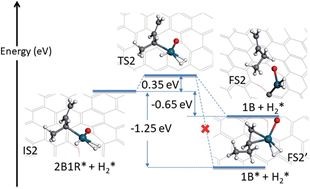
这一新颖的观点在多相催化理论中是十分罕见的,揭示了动力学在多相催化反应中的重要性。这种选择性机制也可能适用于其它单原子催化反应过程,将为单原子异相催化反应提供有效的设计原理。
Feng Y, Zhou L, Wan Q,et al. Selective Hydrogenation of 1, 3-Butadiene Catalyzed by A Single Pd AtomAnchored on Graphene: The Importance of Dynamics[J]. Chemical Science, 2018.
10. DFT计算解释调整d带中心位置如何提升Co4N的HER催化活性(稳定性、自由能、DOS)
中国科学技术大学王功名教授和刘晓静副研究员通过过渡金属掺杂调整了Co4N的d带中心位置,成功赋予其优异的HER催化活性。V掺杂的Co4N纳米片(V-Co4N NS)在10 mA/cm2下碱性介质中的过电势可达37 mV,要远优于Co4N,甚至接近于基准Pt/C催化剂。
在实验的基础上,研究人员进行了DFT模拟计算进一步揭示V掺杂对Co4NHER活性的内在影响。
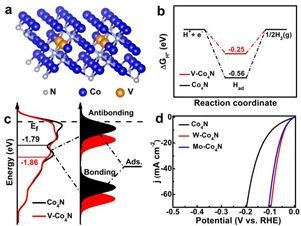
体系能量计算:理论计算表明V掺杂能够降低Co4N体系能量,因而在热力学上是有利的,如图a所示。
自由能计算:图b所示的氢吸收自由能图表明,V-Co4N的吸收H(ΔGH*)为-0.25 eV,比Co4N(-0.56 eV)更接近热力学中性值,这说明V掺杂能够促进氢气的吸附/脱附过程。
DOS计算:费米能级附近的DOS变化可以用于表征吸附物与基体之间的电子相互作用。吸附物价态电子与过渡金属d态电子之间的耦合作用导致形成了分离的成键态和反键态,其中成键态远低费米能级,因而轨道被全部填满,而反键态电子的填充取决于相对于费米能级的能态和键合强度。如图4c所示,Co4N和V-Co4N的d带中心(Ed)为-1.79 eV和-1.85eV,这表明V掺杂后d带中心向更负的方向发生了偏移,反键态的下移弱化了吸附物与基体之间的相互作用,该结果与UPS测试结果相一致。
结合实验结果,研究人员证明通过调整d带中心位置可以有效提升Co4N的HER催化活性。
Chen Z, Song Y, Cai J,et al. Tailoring the d‐Band Centers Enables Co4N Nanosheets To BeHighly Active for Hydrogen Evolution Catalysis[J]. Angewandte ChemieInternational Edition, 2018, 57(18): 5076-5080.
11. 当CP2K遇上催化,OER/ORR理论分析中的绚丽火花!(吸附能、活性位点、自由能)CP2K
CP2K是量子化学和固体物理领域继VASP之后的计算模拟软件新秀。它是由苏黎世大学最新开发的一款免费开源软件,支持力场、半经验方法、HF、MP2、DFT、QM/MM等多种方法,能进行电子结构、CPMD、结构优化、NEB等多种类型的计算。当CP2K遇上催化,会碰撞出炫丽的火花吗?
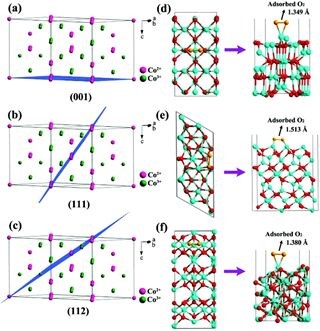
华东师范大学的田阳教授、张立敏副教授(共同通讯作者)团队充分利用MOF材料的结构优势,以MOF的纳米孔结构作为微反应器,从MOF前驱体出发制备出形貌规整、性能优异的OER/ORR双功能电催化剂。
团队用CP2K软件包中的DFT方法对Co9S8@CT催化OER/ORR反应历程中的吸附能及自由能变化进行了计算模拟,结合实验表征与计算结果阐明了Co9S8@CT催化OER/ORR的反应机理,并指明了Co9S8@CT催化剂本征活性的来源。
为了深入挖掘Co9S8@CT-800催化ORR和OER的反应机理,研究人员用CP2K软件包下的DFT方法对Co9S8纳米颗粒在氮掺杂碳纳米管缺陷位点的吸附能、含氧活性物种在催化活性位点的吸附能以及不同电位下OER/ORR反应中的自由能变化进行了计算模拟。
吸附能:DFT计算表明,当Co9S8吸附在碳纳米管表面的N原子空位、C-N原子空位、C原子空位时,整个体系的能量分别上升1.46 eV、2.38 eV、3.36eV,故Co9S8主要吸附在氮掺杂碳纳米管的N原子空位上。
活性位点:为了确定Co9S8@CT-800催化剂中的活性位点,研究人员对含氧活性物种(OOH*、O*、OH*)在Co位点和S位点上的吸附自由能进行了计算模拟,结果表明S位点不能很好的吸附含氧活性物种。而含氧活性物种(OOH*、O*、OH*)在氮掺杂碳纳米管上的吸附能计算表明,含氧活性物种在C位点和N位点上的吸附也不稳定。因此Co9S8@CT-800催化剂的活性位点主要由Co原子提供。
自由能:在不同电位下,Co9S8@CT-800催化OER/ORR的自由能变化曲线会发生显著的变化。
当U=0 V时,吸附在碳纳米管表面N原子空位上的Co9S8在pH为13的碱性电解液中催化ORR的过程是一个放热反应。而U=1.23 V时,形成OOH*需要克服0.31eV的活化能。当U=0.79 V时,这个活化能垒就消失了,此时ORR中的所有基元反应都能在0.44 V的过电位下自发进行,这比Pt理论上需要的过电位(0.45 V)还少。这充分表明Co9S8@CT-800在碱性溶液中的ORR催化活性有望与Pt单质媲美。
对于Co9S8@CT-800在碱性条件下催化的OER,当U=1.23 V时,含氧中间体OOH*的形成步骤需要克服最大的活化能。而当U=1.62 V时,OER中所有的基元反应都变成了下坡反应,此时对应的过电位为0.39 V。这些计算结果都与上述实验结果吻合。总之,无论是OER还是ORR,OOH*的形成都是整个反应的速控步骤。要进一步提升现有催化剂对OER/ORR的双功能电催化活性,就需要从OOH*的形成步骤入手,优化各组分对含氧活性物种的吸附能,在复合催化剂的不同组分之间形成效率更高的协同效应。

Liu T, Zhang L, Tian Y.Earthworm-like N, S-Doped carbon tube-encapsulated Co9S8nanocomposites derived from nanoscaled metal–organic frameworks for highlyefficient bifunctional oxygen catalysis[J]. Journal of Materials Chemistry A,2018, 6(14): 5935-5943. MLA